Neutropenie
Die Liste der möglichen Ursachen einer Neutropenie ist nicht kurz. Die Ätiologie der Neutropenie kann konzeptionell auf zwei Arten betrachtet werden: nach Mechanismus oder ätiologischer Kategorie.
Die Mechanismen, die eine Neutropenie verursachen, sind vielfältig und nicht vollständig geklärt. In vielen Fällen tritt die Neutropenie nach längerer Exposition gegenüber einem Medikament oder einer anderen Substanz auf, was zu einer verminderten Neutrophilenproduktion durch ein hypoplastisches Knochenmark führt. Dies deutet auf eine direkte toxische Wirkung auf die Stammzellen hin. In anderen Fällen ist eine wiederholte, aber intermittierende Exposition gegenüber einem Medikament oder einer anderen Substanz erforderlich. Dies deutet auf einen Immunmechanismus hin, obwohl diese Idee nicht bewiesen ist. In vielen klinischen Situationen sind die genaue Exposition und ihre Dauer in Bezug auf den Beginn der Neutropenie nicht bekannt.
Angesichts dieses unvollständigen Verständnisses der Mechanismen für Neutropenie ist eine Klassifizierung nach einer breiten ätiologischen Kategorie einfacher beizubehalten. In diesem Schema kann die Ätiologie der Neutropenie entweder als angeboren (erblich) oder erworben klassifiziert werden. Auch wenn diese Kategorisierung nur einen begrenzten klinisch-diagnostischen Nutzen hat, kann sie doch nützlich sein, um die erblichen Ursachen der Neutropenie klar von der Vielzahl der erworbenen Ursachen zu trennen. Bei den hereditären Neutropenien können diese Störungen als mit isolierter Neutropenie oder mit anderen Defekten, sei es immunologisch oder phänotypisch, assoziiert beschrieben werden.
Viele hereditäre Störungen sind auf Mutationen in dem Gen zurückzuführen, das für die neutrophile Elastase, ELA2, codiert. Es sind mehrere Allele betroffen. Die häufigsten Mutationen sind intronische Substitutionen, die eine Spleißstelle in Intron 4 inaktivieren. Es sind auch andere Gene als ELA2 betroffen. In der nachstehenden Tabelle sind einige der betroffenen genetischen Bedingungen aufgeführt; es handelt sich um seltene Erkrankungen.
Tabelle 1. Genetische (erbliche) Bedingungen bei Agranulozytose (Tabelle in einem neuen Fenster öffnen)
|
Syndrom |
Inheritance |
Gene |
Clinical Features |
|
Cyclic neutropenia |
Autosomal dominant |
ELA2 |
Alternate 21-day cycling of neutrophils and monocytes |
|
Kostmann syndrome |
Autosomal recessive |
Unknown |
Stable neutropenia, no MDS or AML |
|
Severe congenital neutropenia |
Autosomal dominant |
ELA2 (35-84%) |
Stable neutropenia, MDS or AML |
|
Autosomal dominant |
GFI1 |
Stable neutropenia, circulating myeloid progenitors, lymphopenia |
|
|
Sex linked |
Wasp |
Neutropenic variant of Wiskott-Aldrich syndrome |
|
|
Autosomal dominant |
G-CSFR |
G-CSF–refractory neutropenia, no AML or MDS |
|
|
Hermansky-Pudlak syndrome type 2 |
Autosomal recessive |
AP3B1 |
Severe congenital neutropenia, platelet dense-body defect, oculocutaneous albinism |
|
Chediak-Higashi syndrome |
Autosomal recessive |
LYST |
Neutropenia, oculocutaneous albinism, giant lysosomes, impaired platelet function |
|
Barth syndrome |
Sex linked |
TAZ |
Neutropenia, often cyclic; cardiomyopathy, methylglutaconic aciduria |
|
Cohen syndrome |
Autosomal recessive |
COH1 |
Neutropenia, mental retardation, dysmorphism |
|
Source: Modified from Berliner et al, 2004. AML = acute myeloid leukemia; G-CSF = granulocyte colony-stimulating factor; MDS = myelodysplastic syndrome. |
|||
Die Ursachen für eine erworbene Neutropenie sind vielschichtig, aber die meisten stehen in Zusammenhang mit drei Hauptkategorien: Infektionen, Medikamente (sowohl direkte toxische als auch immunvermittelte) und Autoimmunität. Die chronische benigne Neutropenie oder chronische idiopathische Neutropenie scheint eine Überschneidung mit erblichen und erworbenen Formen zu sein und ist manchmal nicht zu unterscheiden. Einige neutropenische Patienten weisen eine eindeutige Anamnese und ein familiäres Muster auf, während bei anderen Patienten keine Anamnese, nur wenige Bluttests und eine unbekannte Dauer der Neutropenie vorliegen. Bei dieser Patientengruppe könnte eine erbliche oder erworbene Neutropenie vorliegen. Es folgt eine kurze Zusammenfassung der angeborenen und erworbenen neutropenischen Störungen.
Angeborene Neutropenie mit assoziierten Immundefekten
Neutropenie mit abnormalen Immunglobulinen wird bei Personen mit X-chromosomaler Agammaglobulinämie, isoliertem Immunglobulin A (IgA)-Mangel, X-chromosomalem Hyperimmunglobulin M (XHIGM)-Syndrom und Dysgammaglobulinämie Typ I beobachtet. Beim XHIGM-Syndrom, das auf Mutationen im CD40-Liganden zurückzuführen ist, können die Patienten normale oder erhöhte IgM-Spiegel, aber deutlich erniedrigte IgG-Serumspiegel haben. Bei all diesen Erkrankungen ist das Infektionsrisiko hoch, und die Behandlung erfolgt mit intravenösem Immunglobulin (IVIG).
Patienten mit retikulärer Dysgenesie weisen eine schwere Neutropenie, keine zellvermittelte Immunität, Agammaglobulinämie und Lymphopenie auf. Es treten lebensbedrohliche Infektionen auf, die auf den Granulozyten-Kolonie-stimulierenden Faktor (G-CSF) nicht ansprechen. Die Knochenmarktransplantation ist die Behandlung der Wahl.
Kongenitale oder chronische Neutropenien
Schwere kongenitale Neutropenien (SCN) oder Kostmann-Syndrom werden meist rezessiv vererbt und kommen in abgelegenen, isolierten Populationen mit hohem Verwandtschaftsgrad vor. Es wurden auch autosomal dominante und sporadische Fälle gemeldet, die meist auf Mutationen im G-CSF-Rezeptor zurückzuführen sind. Bei diesem Syndrom gibt es keinen einheitlichen Gendefekt. Mutationen in ELA2, die für die zyklische Neutropenie verantwortlich sind (siehe unten), reichen nicht aus, um den Phänotyp der Kostmann-ähnlichen SCN zu erklären.
Die Patienten treten im Alter von 3 Monaten mit wiederkehrenden bakteriellen Infektionen auf. Der Mund und das Perirektum sind die häufigsten Infektionsherde. Diese Art von Neutropenie ist schwerwiegend, und die Behandlung besteht in G-CSF. Das Risiko der Umwandlung in ein myelodysplastisches Syndrom (MDS)/eine akute myeloische Leukämie (AML) mit Monosomie 7 nach G-CSF-Behandlungen ist mit zusätzlichen erworbenen Mutationen verbunden. Die meisten dieser Fälle werden durch eine Mutation im G-CSF-Rezeptor verursacht. Patienten, deren Zustand klinisch auf G-CSF anspricht, werden lebenslang behandelt.
Einige Patienten mit anderen Formen von SCN scheinen Mutationen in GFI1 aufzuweisen, einem Zink-Finger-Transkriptionsrepressor-Gen, das an der Funktion hämatopoetischer Stammzellen und an Entscheidungen über die Abstammung beteiligt ist.
Die zyklische Neutropenie (CN) ist durch periodische Anfälle von Neutropenie in Verbindung mit einer Infektion gekennzeichnet, gefolgt von einer Erholung der peripheren Neutrophilenzahl. Die Periodizität beträgt etwa 21 Tage (Spanne: 12-35 Tage). Die Granulozytenvorläufer verschwinden vor jedem Neutrophilennadir im Zyklus aus dem Knochenmark, weil die Apoptose der myeloischen Vorläuferzellen beschleunigt wird. Einige Fälle können genetisch bedingt sein und werden autosomal rezessiv vererbt. Andere Fälle können auf einen autosomal dominanten Erbgang zurückzuführen sein. In einigen sporadischen Fällen von CN weisen die Patienten Mutationen in ELA2 auf.
Personen mit CN treten typischerweise als Säuglinge oder Kinder auf, es gibt aber auch erworbene Formen von CN im Erwachsenenalter. Die Prognose ist gut, der Verlauf ist gutartig, aber bei 10 % der Patienten kommt es zu lebensbedrohlichen Infektionen. Die Behandlung der zyklischen Neutropenie besteht in täglicher G-CSF-Gabe.
Chronische benigne Neutropenie
Die familiäre chronische benigne Neutropenie oder benigne ethnische Neutropenie ist eine Erkrankung mit autosomal-dominantem Vererbungsmuster, die bei afrikanischer, jemenitisch-jüdischer, äthiopisch-jüdischer, arabischer, karibischer und westindischer Abstammung beobachtet wird. In Populationen afrikanischer und jemenitisch-jüdischer Abstammung zeigen genetische Studien einen starken Zusammenhang mit einem Einzelnukleotid-Polymorphismus im DARC-Gen. Die Patienten sind in der Regel asymptomatisch, und die Infektionen verlaufen mild. Betroffene mit chronischer benigner Neutropenie haben ein insgesamt geringes Infektionsrisiko, und es ist keine spezifische Therapie erforderlich.
Bei nichtfamiliären chronischen benignen Neutropenien sind milde Infektionen mit gutartigem Verlauf typisch für diese Erkrankung. Der ANC reagiert jedoch auf Belastungen wie Infektionen, Kortikosteroide und Katecholamine.
Idiopathische chronische schwere Neutropenie
Die diopathische chronische schwere Neutropenie ist eine Ausschlussdiagnose. Betroffene Patienten weisen Infektionen und schwere Neutropenie auf.
Neutropenie in Verbindung mit phänotypischen Anomalien
Das Shwachman-Syndrom (Shwachman-Diamond) wird autosomal rezessiv vererbt. Die Neutropenie ist mäßig bis schwer mit einer Sterblichkeitsrate von 15-25 %, und das Syndrom zeigt sich im Säuglingsalter mit wiederkehrenden Infektionen, Durchfall und Schwierigkeiten bei der Nahrungsaufnahme. Zwergwuchs, Chondrodysplasie und exokrine Insuffizienz der Bauchspeicheldrüse können auftreten.
Das Shwachman-Diamond-Syndrom und die X-chromosomale Dyskeratosis congenita (DC), die Knorpel-Haar-Hypoplasie (CHH) und die Diamond-Blackfan-Anämie (DBA) scheinen gemeinsame Gendefekte zu haben, die an der Ribosomen-Synthese beteiligt sind. Die meisten Fälle des Shwachman-Diamond-Syndroms werden durch Mutationen im SBDS-Gen verursacht. Die genaue Funktion dieses Gens wird noch erforscht; es ist jedoch an der Ribosomensynthese und an RNA-Verarbeitungsreaktionen beteiligt. Die Behandlung ist G-CSF.
Bei CHH ist das Vererbungsmuster autosomal rezessiv auf Chromosom 9, und es wird in amischen und finnischen Familien beobachtet. CHH wird durch Mutationen im RMRP-Gen verursacht, das für die RNA-Komponente des Ribonuklease-Komplexes zur Verarbeitung mitochondrialer RNA (RNase MRP) kodiert. Die Neutropenie ist mäßig bis schwer. CHH zeigt Defekte der zellvermittelten Immunität, makrozytäre Anämie, gastrointestinale Erkrankungen und Zwergwuchs. Außerdem besteht eine Prädisposition für Krebs, insbesondere für Lymphome. Die Behandlung besteht in einer Knochenmarktransplantation.
Dyskeratosis congenita (Zinsser-Cole-Engman-Syndrom) geht mit geistiger Retardierung, Panzytopenie und defekter zellvermittelter Immunität einher. Dyskeratosis congenita tritt häufiger bei Männern als bei Frauen auf und ähnelt hämatologisch der Fanconi-Anämie. Dyskeratosis congenita ist in der Regel X-chromosomal rezessiv, es gibt jedoch auch autosomal dominante und autosomal rezessive Formen dieser Erkrankung.
Die X-chromosomal rezessive Form der Erkrankung wurde mit Mutationen in DKC1 in Verbindung gebracht, das für Dyskerin kodiert, ein nukleolares Protein, das mit Ribonukleoproteinpartikeln assoziiert ist. Die autosomal dominante Form wird mit Mutationen in einem anderen Gen, TERC, in Verbindung gebracht, das Teil der Telomerase ist. Telomerase hat sowohl eine Protein- als auch eine RNA-Komponente, und TERC kodiert die RNA-Komponente. Patienten mit dieser Störung haben kürzere Telomere als normal. Die Behandlung besteht aus G-CSF, Granulozyten-Makrophagen-Kolonie-stimulierendem Faktor (GM-CSF) und Knochenmarktransplantation.
Das Barth-Syndrom ist eine rezessiv vererbte X-chromosomale Erkrankung mit Kardiomyopathie im Säuglingsalter, Skelettmyopathie, wiederkehrenden Infektionen, Zwergwuchs und mäßiger bis schwerer Neutropenie.
Das Chediak-Higashi-Syndrom ist eine autosomal rezessive Erkrankung mit rezidivierenden Infektionen, geistiger Verlangsamung, Photophobie, Nystagmus, okulokutanem Albinismus, Neuropathie, Blutungsstörungen, Gingivitis und lysosomalen Granula in verschiedenen Zellen. Die Neutropenie ist mäßig bis schwer, und die Behandlung besteht in einer Knochenmarktransplantation.
Myelokathexis
Myelokathexis tritt im Säuglingsalter mit mäßiger Neutropenie auf und ist mit wiederkehrenden Infektionen verbunden. Die Erkrankung ist auf eine beschleunigte Apoptose und eine verminderte Expression von bcl-x in neutrophilen Vorläufern zurückzuführen. Es wird ein abnormales nukleäres Erscheinungsbild beobachtet, mit Hypersegmentierung mit Kernsträngen, Pyknose und zytoplasmatischer Vakuolisierung. Die Behandlung besteht aus G-CSF und GM-CSF.
Lazy Leukozyten-Syndrom
Das Lazy Leukozyten-Syndrom ist eine schwere Neutropenie mit abnormer neutrophiler Motilität. Die Ätiologie ist unbekannt, und die Behandlung ist unterstützend.
Metabolische Störungen
Dabei handelt es sich um chronische Neutropenien mit variablen ANCs. They include glycogen storage disease type 1b and various acidemias, such as isovaleric, propionic, and methylmalonic. In glycogen storage disease type 1b, the treatment is G-CSF and GM-CSF.
Acquired neutropenia caused by intrinsic bone marrow disease
Intrinsic bone marrow diseases that may cause neutropenia include the following:
-
Aplastic anemia
-
Hematologic malignancy (eg, leukemia, lymphoma, myelodysplasia, myeloma)
-
Ionizing radiation
-
Tumor infiltration
-
Granulomatous infection
-
Myelofibrosis
Immune-mediated neutropenia
A drug may act as a hapten and induce antibody formation. This mechanism operates in cases due to gold, aminopyrine, and antithyroid drugs. The antibodies destroy the granulocytes and may not require the continued presence of the drug for their action. Alternatively, the drug may form immune complexes that attach to the neutrophils. This mechanism operates with quinidine.
Drug immune-mediated neutropenia may be caused by the following:
-
Aminopyrine
-
Quinidine
-
Cephalosporins
-
Penicillins
-
Sulfonamides
-
Phenothiazines
-
Hydralazine
Other medications have been implicated
Autoimmune neutropenia is the neutrophil analogue of autoimmune hemolytic anemia and of idiopathic thrombocytopenic neutropenia. It should be considered in the absence of any of the common causes. Antineutrophil antibodies have been demonstrated in these patients. Autoimmune neutropenia may be associated with the following:
-
Rheumatoid arthritis (with or without Felty syndrome)
-
Sjögren syndrome
-
Chronic, autoimmune hepatitis
-
Systemic lupus erythematosus
-
Thymoma
-
Goodpasture disease
-
Granulomatosis with polyangiitis (Wegener granulomatosis)
-
Pure red blood cell (RBC) aplasia, in which there is complete disappearance of granulocyte tissue from the bone marrow; pure RBC dysplasia is a rare disorder due to the presence of antibody-mediated, granulocyte-macrophage colony forming unit (GM-CFU) inhibitory activity, and it is often associated with thymoma
-
Transfusion reactions, which can be caused by the surface antigens of neutrophilia; Empfänger wiederholter Granulozytentransfusionen könnten alloimmunisiert werden
-
Großgranuläre Lymphozytenvermehrung oder Leukämie
Bei der isoimmunen neonatalen Neutropenie produziert die Mutter IgG-Antineutrophilen-Antikörper gegen fetale neutrophile Antigene, die als nicht selbst erkannt werden. Dies kommt bei 3 % der Lebendgeburten vor. Die Störung äußert sich in Form von Neugeborenenfieber, Harnwegsinfektionen, Zellulitis, Lungenentzündung und Sepsis. Die Dauer der Neutropenie beträgt typischerweise 7 Wochen.
Die chronische Autoimmunneutropenie wird bei Erwachsenen beobachtet und hat keine Alterspräferenz. Bis zu 36 % der Patienten weisen antineutrophile Antikörper im Serum auf, und der klinische Verlauf ist in der Regel weniger schwer. Die Patienten können diese Erkrankung in Verbindung mit systemischem Lupus erythematodes, rheumatoider Arthritis, Wegener-Granulomatose und chronischer Hepatitis haben.
Wenn die chronische Autoimmunneutropenie mit diesen Krankheiten in Verbindung steht, sind Kortikosteroide als Behandlung angezeigt. Bei Neugeborenen und Kindern ist diese Erkrankung mit einem geringeren Infektionsrisiko und milderen Infektionen des Mittelohrs, des Magen-Darm-Trakts und der Haut verbunden.
Die T-Gamma-Lymphozytose oder lymphoproliferative Erkrankung ist eine klonale Erkrankung von CD3+ T-Lymphozyten oder CD3- natürlichen Killerzellen (NK), die das Knochenmark und Gewebe infiltrieren. Die auch als Leukämie der großkörnigen Lymphozyten (LGL-Leukämie) bekannte T-Gamma-Lymphozytose kann mit rheumatoider Arthritis einhergehen und ist mit hochtitrigen antineutrophilen Antikörpern verbunden. Die Neutropenie ist anhaltend und schwerwiegend. Die Behandlung ist oft unterstützend, zielt aber auch darauf ab, die klonale Population zu eliminieren.
Erworbene Neutropenie durch Infektionen
Infektionen sind die häufigste Form der erworbenen Neutropenie. Zu den Infektionen, die eine Neutropenie verursachen können, gehören unter anderem die folgenden:
-
Bacterial sepsis
-
Viral infections (eg, influenza, measles, Epstein Barr virus , cytomegalovirus , viral hepatitis, human immunodeficiency virus -1) (see first image below)
-
Toxoplasmosis
-
Brucellosis
-
Typhoid
-
Tuberculosis (see second and third images below)
-
Malaria
-
Dengue fever
-
Rickettsial infection
-
Babesiosis
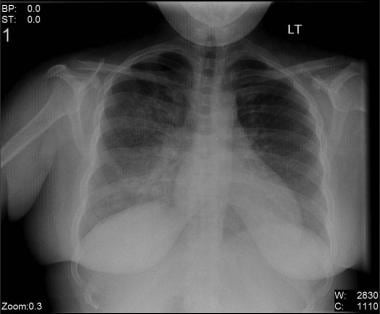 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia. 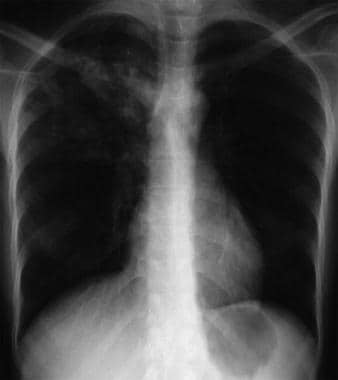 Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Die Frau wurde isoliert aufgenommen und erhielt in der Notaufnahme empirisch ein 4-Medikamenten-Regime. Die Tuberkulose wurde durch einen Sputumtest bestätigt. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Die Frau wurde isoliert aufgenommen und erhielt in der Notaufnahme empirisch ein 4-Medikamenten-Regime. Die Tuberkulose wurde durch einen Sputumtest bestätigt. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org. 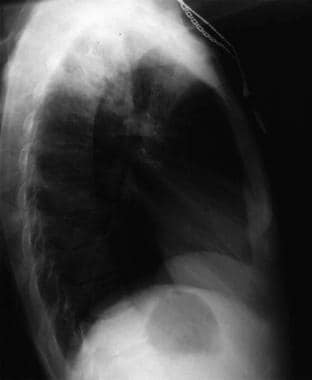 Seitliche Röntgenaufnahme des Brustkorbs bei einem 31-jährigen Patienten mit Influenza-Pneumonie. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org.
Seitliche Röntgenaufnahme des Brustkorbs bei einem 31-jährigen Patienten mit Influenza-Pneumonie. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org.
 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.  Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Die Frau wurde isoliert aufgenommen und erhielt in der Notaufnahme empirisch ein 4-Medikamenten-Regime. Die Tuberkulose wurde durch einen Sputumtest bestätigt. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Die Frau wurde isoliert aufgenommen und erhielt in der Notaufnahme empirisch ein 4-Medikamenten-Regime. Die Tuberkulose wurde durch einen Sputumtest bestätigt. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org.  Seitliche Röntgenaufnahme des Brustkorbs bei einem 31-jährigen Patienten mit Influenza-Pneumonie. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org.
Seitliche Röntgenaufnahme des Brustkorbs bei einem 31-jährigen Patienten mit Influenza-Pneumonie. Bild mit freundlicher Genehmigung von Remote Medicine, remotemedicine.org. Die am häufigsten beteiligten Organismen stammen aus der endogenen Flora. Staphylococcus aureus-Organismen werden bei Hautinfektionen gefunden. Gramnegative Organismen werden bei Infektionen der Harnwege und des Magen-Darm-Trakts beobachtet, insbesondere Escherichia coli und Pseudomonas-Arten. Auch Candida albicans-Infektionen können auftreten. In der Mundhöhle kann eine gemischte Flora gefunden werden.
Virale Infektionen führen häufig zu einer leichten oder mäßigen Neutropenie. Eine Agranulozytose ist selten, kann aber auftreten. Die häufigsten Erreger sind das Epstein-Barr-Virus, das Hepatitis-B-Virus, das Gelbfiebervirus, das Cytomegalovirus und die Influenza. Viele überwältigende Infektionen, sowohl virale als auch bakterielle, können eine schwere Neutropenie verursachen.
Erworbene Neutropenie durch Ernährungsmangel
Zu den Ernährungsmängeln, die eine Neutropenie verursachen können, gehören Vitamin B-12, Folsäure und Kupfermangel.
Erworbene Neutropenie durch Medikamente und Chemikalien, ausgenommen zytotoxische Chemotherapie
Zahlreiche Medikamente wurden mit Neutropenie in Verbindung gebracht. Die höchsten Risikokategorien sind Schilddrüsenmedikamente, Makrolide und Procainamide. Wie bereits erwähnt, wirken viele Medikamente über einen immunvermittelten Mechanismus. Einige Medikamente scheinen jedoch direkte toxische Wirkungen auf Knochenmarkstammzellen oder neutrophile Vorläuferzellen im mitotischen Kompartiment zu haben. So können beispielsweise Medikamente wie Antipsychotika und Antidepressiva sowie Chloramphenicol bei einigen Personen aufgrund ihres Stoffwechsels und ihrer Empfindlichkeit auf diese Weise als direkte Toxine wirken. Andere Medikamente können eine Kombination aus immunologischen und nicht-immunologischen Mechanismen aufweisen oder unbekannte Wirkmechanismen haben.
Antimikrobielle Mittel sind Penicillin, Cephalosporine, Vancomycin, Chloramphenicol, Gentamicin, Clindamycin, Doxycyclin, Flucytosin, Nitrofurantoin, Novobiocin, Minocyclin, Griseofulvin, Lincomycin, Metronidazol, Rifampin, Isoniazid, Streptomycin, Thiacetazon, Mebendazol, Pyrimethamin, Levamisol, Ristocetin, Sulfonamide, Chloroquin, Hydroxychloroquin, Chinacrin, Ethambutol, Dapson, Ciprofloxacin, Trimethoprim, Imipenem/Cilastatin, Zidovudin, Fludarabin, Acyclovir und Terbinafin.
Analgetika und entzündungshemmende Mittel umfassen Aminopyrin, Dipyron, Indomethacin, Ibuprofen, Acetylsalicylsäure, Diflunisal, Sulindac, Tolmetin, Benoxaprofen, Barbiturate, Mesalazin und Chinin.
Antipsychotika, Antidepressiva und neuropharmakologische Wirkstoffe umfassen Phenothiazine (Chlorpromazin, Methylpromazin, Mepazin, Promazin, Thioridazin, Prochlorperazin, Trifluoperazin, Trimeprazin), Clozapin, Risperidon, Imipramin, Desipramin, Diazepam, Chlordiazepoxid, Amoxapin, Meprobamat, Thiothixen und Haloperidol.
Zu den Antikonvulsiva gehören Valproinsäure, Phenytoin, Trimethadion, Mephenytoin (Mesantoin), Ethosuximid und Carbamazepin.
Zu den Antithyreostatika gehören Thiouracil, Propylthiouracil, Methimazol, Carbimazol, Kaliumperchlorat und Thiocyanat.
Kardiovaskuläre Arzneimittel umfassen Procainamid, Captopril, Aprindin, Propranolol, Hydralazin, Methyldopa, Chinidin, Diazoxid, Nifedipin, Propafenon, Ticlopidin und Vesnarinon.
Antihistaminika umfassen Cimetidin, Ranitidin, Tripelennamin (Pyribenzamin), Methaphenilen, Thenalidin, Brompheniramin und Mianserin.
Diuretika umfassen Acetazolamid, Bumetanid, Chlorothiazid, Hydrochlorothiazid, Chlorthalidon, Methazolamid und Spironolacton.
Blutzuckermittel sind Chlorpropamid und Tolbutamid.
Antimalariamittel sind Amodiaquin, Dapson, Hydroxychloroquin, Pyrimethamin und Chinin.
Zu den sonstigen Arzneimitteln zählen Allopurinol, Colchicin, Aminoglutethimid, Famotidin, Bezafibrat, Flutamid, Tamoxifen, Penicillamin, Retinsäure, Metoclopramid, Phenindion, Dinitrophenol, Ethacrynsäure, Dichlordiphenyltrichlorethan (DDT), Cinchophen, Antimon, Pyrithyldion, Ruwolfia, Ethanol, Chlorpropamid, Tolbutamid, Thiazide, Spironolacton, Methazolamid, Acetazolamid, IVIG und Levodopa.
Zu den Schwermetallen gehören Gold, Arsen und Quecksilber.
Die häufigste Ursache der Agranulozytose ist die Exposition gegenüber Medikamenten oder Chemikalien: Bei etwa der Hälfte der Patienten liegt eine medikamentöse oder chemische Exposition vor. Jede Chemikalie oder jedes Medikament, das das Knochenmark deprimieren und eine Hypoplasie oder Aplasie verursachen kann, ist in der Lage, eine Agranulozytose zu verursachen. Bei einigen Medikamenten ist dies bei jedem der Fall, wenn sie in ausreichend hohen Dosen verabreicht werden. Andere Wirkstoffe scheinen idiosynkratische Reaktionen hervorzurufen, die nur bestimmte empfängliche Personen betreffen.
Einige Wirkstoffe (z.B. Valproinsäure, Carbamazepin und Beta-Lactam-Antibiotika) wirken durch direkte Hemmung der Myelopoese. In Knochenmarkskulturen hemmen diese Wirkstoffe dosisabhängig die Bildung von Granulozytenkolonien. In den meisten anderen Fällen spielt eine direkte Schädigung der Mikroumgebung des Knochenmarks oder der myeloischen Vorläufer eine Rolle.
Viele Medikamente, die mit Agranulozytose in Verbindung gebracht werden, wurden der US-amerikanischen Food and Drug Administration (FDA) im Rahmen ihrer Meldepflicht für unerwünschte Reaktionen gemeldet. Viele Wirkstoffe werden auch an ein von der American Medical Association (AMA) geführtes Register gemeldet. Die gemeldeten Arzneimittel wurden allein, in Kombination mit einem anderen Arzneimittel, das als potenziell toxisch bekannt ist, oder mit einem anderen Arzneimittel ohne bekannte Toxizität verwendet. Mehrere Arzneimittel sind besonders hervorzuheben, da sie sehr häufig mit Agranulozytose in Verbindung gebracht werden. Dazu gehören die folgenden:
-
Phenothiazine
-
Antithyroid drugs (thiouracil and propylthiouracil)
-
Aminopyrine
-
Chloramphenicol
-
Sulfonamides
Miscellaneous immunologic neutropenias
Immunologic neutropenias may occur after bone marrow transplantation and blood product transfusions.
Felty syndrome is a syndrome of rheumatoid arthritis, splenomegaly, and neutropenia. Splenectomy shows an initial response, but neutropenia may recur in 10-20% of patients. Treatment is directed toward rheumatoid arthritis.
In complement activation–mediated neutropenia, hemodialysis, cardiopulmonary bypass, and extracorporeal membrane oxygenation (ECMO) expose blood to artificial membranes and can cause complement activation with subsequent neutropenia.
In splenic sequestration, the degree of neutropenia resulting from this process is proportional to the severity of the splenomegaly and the bone marrow’s ability to compensate for the reduction in circulating bands and neutrophils.
Eosinopenia and basophilopenia
Eosinopenia may be associated with the following:
-
Acute bacterial infection
-
Glucocorticoid administration
-
Physical stress
-
Thymoma
Decreased circulating basophils may be associated with the following:
-
Anaphylaxis
-
Acute infection
-
Drug-induced hypersensitivity
-
Congenital absence of basophils
-
Hemorrhage
-
Hyperthyroidism
-
Ionizing radiation
-
Neoplasia
-
Ovulation
-
Urticaria
-
Drugs (eg, corticosteroid, adrenocorticotropic hormone therapy, chemotherapeutic agents, thyroid hormones)
Go to Pediatric Autoimmune and Chronic Benign Neutropenia for complete information on this topic.