Determinación de la mutación V617F del gen JAK2 en los síndromes mieloproliferativos crónicos en nuestro país: a propósito de un caso
Determinación de la mutación V617F del gen JAK2 en los síndromes mieloproliferativos crónicos en nuestro país: a propósito de un caso
Dres. Daniela Lens*, Pablo Muxi†, Lics. Andreína Brugnini‡,
Natalia Trías‡, Dra. Silvia Pierri§
Hospital de Clínicas. Facultad de Medicina.
Universidad de la República. Montevideo, Uruguay
Resumen
La policitemia vera (PV), la trombocitemia esencial (TE) y la mielofibrosis idiopática (MI) son trastornos mieloproliferativos clonales estrechamente relacionados y caracterizados por una proliferación excesiva de una o más líneas mieloides tales como eritrocitos, plaquetas y fibroblastos de la médula ósea.
Si bien existen estrictos criterios para el diagnóstico de estos síndromes mieloproliferativos, la categorización precisa continúa siendo un objeto de debate y adicionalmente estos desórdenes son difíciles de diferenciar de procesos reactivos en muchas ocasiones.
Recientemente, en el año 2005, se identificó en varias de estas entidades una mutación en el gen tirosina quinasa Janus kinase 2 (JAK2). Esta mutación consiste en la sustitución de una G por una T en la posición 1849, resultando en la sustitución en la proteína de una fenilalanina por valina (JAK2 V617F).
La incidencia de esta mutación se observó en cerca de 90% de los casos con PV y en aproximadamente 50% de los casos con MI y TE.
En este trabajo describiremos la detección de esta mutación en un paciente con diagnóstico de probable PV mediante un ensayo de reacción en cadena de la polimerasa (PCR) alelo específica de alta sensibilidad para la detección de esta mutación y discutiremos la importancia de esta mutación de descubrimiento reciente en el diagnóstico y tratamiento de los síndromes mieloproliferativos BCR-ABL negativos.
Palabras clave: TRASTORNOS MIELOPROLIFERATIVOS – diagnóstico.
MUTACION- genética.
PROTEINA-TIROSINA QUINASA.
* Prof. Agregada. Departamento Básico de Medicina.
† Prof. Agregado. Clínica Hematológica. Departamento Clínico de Medicina.
‡ Lic. Bioquímica. Ayudante. Departamento Básico de Medicina.
§ Prof. Adjunta. Clínica Hematológica. Departamento Clínico de Medicina.
Correspondencia: Dra. Daniela Lens.
Depto. Básico de Medicina. Hospital de Clínicas. Piso 15. Avda Italia s/n. Montevideo CP 11600. Montevideo, Uruguay.
Correo electrónico: [email protected]
Recibido: 31/7/06.
Aceptado: 26/2/07.
Introducción
A diferencia de lo que ocurre en la leucemia mieloide crónica donde la identificación del rearreglo específico BCR-ABL condujo a un gran avance en el diagnóstico, seguimiento y tratamiento de esta entidad, en los síndromes mieloproliferativos crónicos (SMP) BCR-ABL negativos, tales como la policitemia vera (PV), trombocitemia esencial (TE) y mielofibrosis idiopática (MI), no se conocían hasta muy recientemente alteraciones genéticas específicas.
Entre mayo y junio de 2005, cinco grupos de investigadores describieron una nueva mutación puntual del gen tirosina quinasa JAK2, debida a la sustitución de una guanina por una timina en el nucleótido 1849 del exón 14, lo cual resulta en el reemplazo de una valina por una fenilalanina en la posición 617 de la proteína, por la cual se denomina a la mutación JAK2 V617F(1-5).
Esta mutación se observó en cerca de 90% de los casos con PV cuando se usan ensayos de alta sensibilidad para detectarla(6). También se encontró en aproximadamente 50% de los casos con MI y TE, mientras que no se ha encontrado en sujetos sanos ni en pacientes con eritrocitosis secundaria, por lo cual esta mutación tiene un muy alto valor predictivo en distinguir SMPC de condiciones no clonales tales como las policitemias secundarias.
Esta mutación ocurre en una región altamente conservada de un dominio autoinhibitorio que regula negativamente la señalización del JAK2. Diversos trabajos han demostrado que esta mutación participa en la patogenia de estas condiciones, especialmente debido a una ganancia de función del gen JAK2 y mediante una pérdida de control que se asocian con la excesiva mieloproliferación de estos desórdenes.
En este trabajo describiremos la detección de esta mutación en un paciente con diagnóstico de probable PV y discutiremos la importancia de esta nueva mutación en el diagnóstico y tratamiento de los SMP BCR-ABL negativos.
Material y método
Extracción de ácido desoxirribonucleico
La extracción de ácido desoxirribonucleico (ADN) genómico se realizó a partir de 1 ml de sangre periférica citratada, utilizando el reactivo comercial dnazol (Life Techno-logies) y siguiendo el protocolo indicado por el fabricante. Las muestras de ADN fueron conservadas a 4°C.
Detección de la mutación Vl617F
La mutación V617F se detectó a partir del ADN genómico previamente obtenido, mediante reacción en cadena de la polimerasa (PCR) alelo específica, según el método publicado por Baxter y colaboradores(1). Esta PCR está diseñada de forma que se utilizan tres cebadores diferentes, denominados JAK2R, JAK2F/IC y JAK2F (tabla 1). El cebador JAK2F es específico para el alelo mutante y el cebador JAK2F/IC se encuentra en todos los individuos independientemente de la presencia o no de la mutación, por lo que constituye un control interno de la reacción. De esta forma, en todos los individuos se amplifica un producto de 364 pares de bases (pb) con los cebadores JAK2F/IC y JAK2R, y solamente en los individuos que presentan la mutación en estudio se obtiene un producto de 203 pb utilizando los cebadores JAK2F y JAK2R (figura 1).
La amplificación por PCR fue realizada en un volumen final de 20 uL utilizando 500 ng de ADN, 200 mM/L de cada dNTPs, 0,5 U de Taq polimerasa (New England Biolabs) y 2 uL de buffer Taq 10x conteniendo 2mM de MgCl (New England Biolabs), 0,5 mM de los cebadores JAK2 F y JAK2 F/IC y 1 mM del cebador JAK2 R.
El programa de PCR utilizado consta de 36 ciclos, y se inicia con una desnaturalización de 5 minutos, para luego ciclar: 30 segundos a 94°C, 30 segundos a 58°C y 30 segundos a 72°C, terminando con una extensión final de 5 minutos. Se incluyó siempre en todas las reacciones un control negativo que contiene todos los reactivos necesarios para la amplificación y donde se sustituye el molde ADN por agua. La verificación del producto de PCR esperado se realizó en un gel de agarosa a 2% teñido con bromuro de etidio(7).

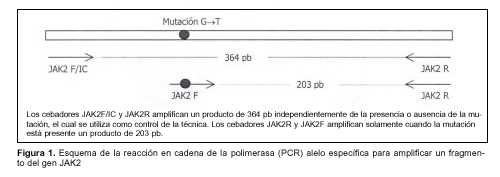
Análisis del caso
Paciente de sexo masculino, 72 años, fumador intenso, con hipertensión arterial crónica en tratamiento y portador de una arteriopatía obstructiva crónica que en el año 2002 determina una amputación del quinto dedo del pie izquierdo. En diciembre de 2004, presentando un nuevo episodio obstructivo de miembro inferior izquierdo, mostró en su hemograma un aumento de las cifras de eritrocitos con un hematocrito de 66%, conjuntamente a una granulocitosis de 24.000 y plaquetas de 245.000. En la evolución se mantienen las cifras de hematocrito aumentando progresivamente las cifras de plaquetas hasta 710.000. En el estudio de biopsia de médula ósea se observó una médula ósea hipercelular donde destaca una hiperplasia de la serie eritroide, por lo cual concluyen en un síndrome mieloproliferativo crónico tipo PV.
El paciente es tratado con flebotomías y en mayo de 2005 se inicia tratamiento con hidroxiurea, el cual se suspende en febrero de 2006 por una complicación hemorrágica.
Como se ilustra en la figura 2, el paciente mostró en el estudio de PCR alelo específico para la mutación V617F del gen JAK2 un patrón con dos bandas. Por un lado, la banda control de 364 pb que también está presente en los dos controles sanos que se estudiaron. Por otro, se amplificó también en el paciente un producto de 203 pb demostrando la presencia de la mutación. Esta banda no se observó en los controles sanos.
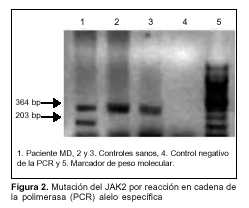
Discusión y conclusiones
En este trabajo hemos descripto el primer caso en el cual se ha identificado la mutación V617 del gen JAK2 en nuestro país. Este trabajo se enmarca en un proyecto multidisciplinario entre el Departamento Básico de Medicina y el Servicio de Hematología del Hospital de Clínicas, con vistas a determinar la presencia de esta mutación en 100 pacientes con SMP BCR-ABL negativo.
La identificación reciente de la mutación activadora V617F en el gen JAK2 representa un importante avance en el conocimiento de la patogenia de los síndromes mieloproliferativos BCR-ABL negativos. Debido a la alta frecuencia en que esta mutación es encontrada en la PV y en menor grado en la TE y la MI, se ha postulado que la detección de esta mutación tendría un fuerte impacto en el diagnóstico, la clasificación y el tratamiento de estas entidades. Distintos grupos han propuesto incorporar la detección de esta mutación como un test diagnóstico de primera línea cuando nos encontramos con un hematocrito mayor a 51% o frente a sospecha de PV, dado que la detección del JAK2 V617F tiene 100% de valor predictivo positivo del diagnóstico de PV(8,9). Por otra parte, distintos autores han postulado que la presencia de esta mutación permitiría una nueva clasificación de los SMP, ya que las TE, PV y MI portadoras de la mutación JAK2 podrían representar la misma enfermedad en distintas etapas evolutivas en lugar de distintas entidades(10-12). De todos modos, el valor clínico exacto y el beneficio de la detección de esta mutación está por establecerse y necesita de ensayos clínicos prospectivos especialmente diseñados para resolver estas preguntas.
Adicionalmente, el descubrimiento de esta mutación abre nuevas perspectivas para terapias dirigidas específicamente a la proteína quinasa mutada, en forma similar a los inhibidores del BCR-ABL en las leucemias mieloides crónicas.
Summary
Polycythemia vera (PV), essential thrombocythemia (ET–TE) and idiopathic myelofibrosis (IM-MI) are clonal myeloproliferative disorders characterized by an excessive proliferation of one or more myeloids lineage such as erythrocits, platelets and fibroblasts of bone marrow.
Precise categorization of myeloproliferative syndromes still need to be debated even if diagnostic criteria are strict; additionally these disorders are difficult to differentiate from reactive processes.
Recently, in 2005, JAK2-mutation was identified in many of those entities. Sequencing of the coding region of JAK2 revealed a G to T transversion at position 1849, that changed a valine to a phenylalanine (JAK2 V617F).
Incidence of V617F-JAK2-mutation was almost 90% in patients with PV, and 50% in patients with IM and ET.
In this study we describe the detection of the V617F-JAK2-mutation in a patient suspected of PV using allele-specific polymerase chain reaction analysis (PCR) and we discuss the importance of the mutation for diagnosis and treatment of negative BCR-ABL myeloproliferative syndromes.
Résumé
La polycytémie vera (PV), La trombocitemia esencial (TE) y la mielofibrosis idiopática (MI) son trastornos mieloproliferativos clonales estrechamente relacionados que se caracterizan por la proliferación excesiva de una o más líneas mieloides como los eritrocitos, las plaquetas y los fibroblastos de la médula ósea.
Aunque existen criterios estrictos para el diagnóstico de estos síndromes mieloproliferativos, la categorización precisa sigue siendo objeto de debate e, incluso, estos trastornos son difíciles de diferenciar de los procesos reactivos en varias ocasiones.
Recientemente, en 2005, se identificó una mutación en el gen de la tirosina quinasa Janus quinasa 2 (JAK2) en varias de estas entidades. Esta mutación consiste en la sustitución de una G por una T en la posición 1849, dando lugar a la sustitución en la proteína de una fenilalanina por valina (JAK2 V617F).
La incidencia de esta mutación se ha observado en casi el 90% de los casos con PV y, en aproximadamente el 50% de los casos con MI y TE.
En este trabajo describiremos la detección de esta mutación en un paciente con diagnóstico de probable PV mediante un ensayo de reacción en cadena de la polimerasa (PCR) específico para el alelo con alta sensibilidad para la detección de esta mutación y discutiremos la importancia de esta mutación recién descubierta en el diagnóstico y tratamiento de BCR-Síndromes mieloproliferativos ABL negativos.
Resumen
La Policitemia Vera (PV), la trombocitemia hemorrágica (TE) y la mielofibrosis idiopática (MI) son trastornos mieloproliferativos clónicos fuertemente relacionados y caracterizados por una proliferación excesiva de una o más líneas mielóides como eritrocitos, plaquetas y fibroblastos de la médula ósea.
Aunque existen criterios rigurosos para el diagnóstico de estos síndromes mieloproliferativos, la clasificación precisa sigue siendo discutida; además, muchas veces es muy difícil diferenciar estos distritos de procesos reativos.
En 2005, se identificó una mutación en el gen tirosina quinasa Janus quinasa 2 (JAK2) en varias de estas entidades. En esta mutación se observa una sustitución de G a T en la posición 1849, que conduce a una sustitución de fenilalanina a valina (JAK2 V617F) en la proteína.
Esta mutación se observó en aproximadamente el 90% de los casos con PV y en aproximadamente el 50% de los casos con MI y TE.
En este trabajo describimos la detección de esta mutación en un paciente con un diagnóstico probable de PV utilizando un ensayo de reacción en cadena de la polimerasa (PCR) altamente sensible y específico para el alelo para la detección de esta mutación y discutimos la importancia de esta mutación recién descubierta en el diagnóstico y tratamiento de los síndromes mieloproliferativos BCR-ABL-negativos.
Bibliografía
1. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054-61.
2. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Layout C, et al. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythaemia vera. Nature 2005; 434: 1144-8.
3. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain of function mutation in JAK2 is frequently found in patients with myeloproliferative disorders. N Engl J Med 2005; 352: 1779-90.
4. Levine RL, Waldleigh M, Cools J, Ebert BL, Werning G, Huntly BJ, et al. Mutación activadora en la tirosina quinasa JAK2 en policitemia vera, trombocitemia esencial y metaplasia mieloide con mielofibrosis. Cancer Cell 2005; 7: 387-97.
6. Vainshenker W, Constantinescu SN. Una mutación de activación única en JAK2 (V617F) está en el origen de la policitemia vera y permite una nueva clasificación de las enfermedades mieloproliferativas. Hematology Am Soc Hematol Educ Program 2005; 195-200.
7. Sambrook J, Russell DW. Clonación molecular: un manual de laboratorio. 3 ed. New York: Cold Spring Harbor Laboratory, 2001.
8. Tefferi A, Pardanani A. Cribado de mutaciones para JAK2V617F: cuándo pedir la prueba y cómo interpretar los resultados. Leuk Res 2006; 30(6): 739-44.
9. James C, Delhommeau F, Marzac C, Teyssandier I, Couedic JP, Giraudier S, et al. Detección de JAK2 V617F como prueba diagnóstica de primera intención para la eritrocitosis. Leukemia 2006; 20: 350-3.
10. Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, et al. Definición de subtipos de trombocitemia esencial y relación con la policitemia vera basada en el estado de la mutación JAK2 V617F: un estudio prospectivo. Lancet 2005; 366: 1945-53.
11. Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, Ponziani V, et al. Implicaciones clínicas de la mutación JAK2 V617F en la trombocitemia esencial. Leukemia 2005; 19: 1847-9.