Neutropenia
La lista de todas las causas potenciales de neutropenia no es corta. La etiología de la neutropenia puede verse conceptualmente de dos maneras amplias, por mecanismo o por categoría etiológica.
Los mecanismos que causan la neutropenia son variados y no se entienden completamente. En muchos casos, la neutropenia se produce tras una exposición prolongada a un fármaco u otra sustancia, lo que provoca una disminución de la producción de neutrófilos por parte de la médula ósea hipoplásica. Esto sugiere un efecto tóxico directo de las células madre. En otros casos, es necesaria una exposición repetida pero intermitente al fármaco u otra sustancia. Esto sugiere un mecanismo inmunológico, aunque esta idea no ha sido probada. En muchas situaciones clínicas, se desconoce la exposición exacta y su duración en relación con la aparición de la neutropenia.
En vista de esta comprensión incompleta de los mecanismos de la neutropenia, es más sencillo mantener la clasificación por categorías etiológicas amplias. En este esquema, la etiología de la neutropenia puede clasificarse como congénita (hereditaria) o adquirida. Aunque esta categorización puede tener una utilidad diagnóstica clínica limitada, puede ser útil para separar claramente las causas hereditarias de neutropenia de la panoplia de causas adquiridas. En el contexto de las neutropenias hereditarias, estos trastornos pueden describirse además como asociados a una neutropenia aislada o a otros defectos, ya sean inmunológicos o fenotípicos.
Muchos trastornos hereditarios se deben a mutaciones en el gen que codifica la elastasa de neutrófilos, ELA2. Hay varios alelos implicados. Las mutaciones más comunes son sustituciones intrónicas que inactivan un sitio de empalme en el intrón 4. También están implicados otros genes además del ELA2. La siguiente tabla enumera algunas de las condiciones genéticas implicadas; se trata de condiciones poco comunes.
Tabla 1. Condiciones genéticas (hereditarias) en la agranulocitosis (Abrir tabla en una ventana nueva)
|
Síndrome |
Inheritance |
Gene |
Clinical Features |
|
Cyclic neutropenia |
Autosomal dominant |
ELA2 |
Alternate 21-day cycling of neutrophils and monocytes |
|
Kostmann syndrome |
Autosomal recessive |
Unknown |
Stable neutropenia, no MDS or AML |
|
Severe congenital neutropenia |
Autosomal dominant |
ELA2 (35-84%) |
Stable neutropenia, MDS or AML |
|
Autosomal dominant |
GFI1 |
Stable neutropenia, circulating myeloid progenitors, lymphopenia |
|
|
Sex linked |
Wasp |
Neutropenic variant of Wiskott-Aldrich syndrome |
|
|
Autosomal dominant |
G-CSFR |
G-CSF–refractory neutropenia, no AML or MDS |
|
|
Hermansky-Pudlak syndrome type 2 |
Autosomal recessive |
AP3B1 |
Severe congenital neutropenia, platelet dense-body defect, oculocutaneous albinism |
|
Chediak-Higashi syndrome |
Autosomal recessive |
LYST |
Neutropenia, oculocutaneous albinism, giant lysosomes, impaired platelet function |
|
Barth syndrome |
Sex linked |
TAZ |
Neutropenia, often cyclic; cardiomyopathy, methylglutaconic aciduria |
|
Cohen syndrome |
Autosomal recessive |
COH1 |
Neutropenia, mental retardation, dysmorphism |
|
Source: Modified from Berliner et al, 2004. AML = acute myeloid leukemia; G-CSF = granulocyte colony-stimulating factor; MDS = myelodysplastic syndrome. |
|||
Las causas de la neutropenia adquirida son complejas, pero la mayoría están relacionadas con tres categorías principales: infección, fármacos (tanto tóxicos directos como inmunomediados) y autoinmunidad. La neutropenia crónica benigna, o neutropenia crónica idiopática, parece ser un trastorno que se solapa con las formas hereditarias y adquiridas, y a veces es indistinguible. Algunos pacientes neutropénicos presentan una historia clara y un patrón familiar, mientras que otros carecen de antecedentes familiares, tienen pocas determinaciones analíticas y una duración desconocida de la neutropenia. Este grupo de pacientes podría tener una neutropenia hereditaria o adquirida. A continuación se presenta un breve resumen de los trastornos neutropénicos congénitos y adquiridos.
Nutropenia congénita con defectos inmunitarios asociados
La neutropenia con inmunoglobulinas anormales se observa en individuos con agammaglobulinemia ligada al cromosoma X, deficiencia aislada de inmunoglobulina A (IgA), síndrome de hiperinmunoglobulina M ligado al cromosoma X (XHIGM) y disgammaglobulinemia tipo I. En el XHIGM, que se debe a mutaciones en el ligando CD40, los pacientes pueden tener niveles normales o elevados de IgM pero niveles séricos de IgG notablemente reducidos. En todos estos trastornos, el riesgo de infección es elevado y el tratamiento es la inmunoglobulina intravenosa (IGIV).
Los pacientes con disgenesia reticular presentan neutropenia grave, ausencia de inmunidad celular, agammaglobulinemia y linfopenia. Se producen infecciones potencialmente mortales que son refractarias al factor estimulante de colonias de granulocitos (G-CSF). El trasplante de médula ósea es el tratamiento de elección.
Neutropenias congénitas o crónicas
La neutropenia congénita severa (NSC), o síndrome de Kostmann, suele ser causada por una herencia recesiva y se encuentra en poblaciones remotas y aisladas con un alto grado de consanguinidad. También se han descrito casos autosómicos dominantes y esporádicos, la mayoría debidos a mutaciones en el receptor del G-CSF. No existe un defecto genético uniforme en este síndrome. Las mutaciones en ELA2, que son causantes de la neutropenia cíclica (véase más adelante) no son suficientes para explicar el fenotipo del SCN tipo Kostmann.
Los pacientes se presentan a la edad de 3 meses con infecciones bacterianas recurrentes. La boca y el perirrectal son los sitios más comunes de infección. Este tipo de neutropenia es grave y el tratamiento es G-CSF. El riesgo de conversión a síndrome mielodisplásico (SMD)/leucemia mielógena aguda (LMA) con monosomía 7 después de los tratamientos con G-CSF está asociado a mutaciones adicionales adquiridas. La mayoría de estos casos están causados por una mutación en el receptor del G-CSF. Los pacientes cuya afección responde clínicamente al G-CSF son tratados de por vida.
Algunos pacientes con otras formas de SCN parecen tener mutaciones en GFI1, un gen represor transcripcional de dedos de zinc que interviene en la función de las células madre hematopoyéticas y en las decisiones de compromiso de linaje.
La neutropenia cíclica (NC) se caracteriza por brotes periódicos de neutropenia asociados a infecciones, seguidos de una recuperación del recuento de neutrófilos periféricos. Su periodicidad es de unos 21 días (rango, 12-35 d). Los precursores de granulocitos desaparecen de la médula antes de cada nadir de neutrófilos en el ciclo debido a la apoptosis acelerada de las células progenitoras mieloides. Algunos casos pueden estar determinados genéticamente con una herencia autosómica recesiva. Otros casos pueden deberse a una herencia autosómica dominante. En algunos casos esporádicos de CN, los pacientes tienen mutaciones en ELA2.
Las personas con CN suelen presentarse como bebés o niños, pero existen formas adquiridas de CN en la edad adulta. El pronóstico es bueno, con un curso benigno; sin embargo, el 10% de los pacientes experimentan infecciones que ponen en peligro su vida. El tratamiento para la neutropenia cíclica es G-CSF diario.
Neutropenia crónica benigna
La neutropenia crónica benigna familiar, o neutropenia étnica benigna, es un trastorno con un patrón de herencia autosómico dominante observado en descendientes de africanos, judíos yemenitas, judíos etíopes, árabes, caribeños y antillanos. En las poblaciones de ascendencia africana y judía yemenita, los estudios genéticos muestran una fuerte asociación con un polimorfismo de un solo nucleótido en el gen DARC. Los pacientes suelen ser asintomáticos y las infecciones son leves. Los individuos afectados con neutropenia crónica benigna tienen un riesgo general bajo de infección y no se requiere una terapia específica.
En las neutropenias crónicas benignas no familiares, las infecciones leves con un curso benigno tipifican este trastorno. El CNA, sin embargo, sí responde al estrés, como la infección, los corticosteroides y las catecolaminas.
Nutropenia crónica grave idiopática
La neutropenia crónica grave idiopática es un diagnóstico de exclusión. Los pacientes afectados presentan infecciones y neutropenia grave.
Neutropenia asociada a anomalías fenotípicas
El síndrome de Shwachman (Shwachman-Diamond) tiene un patrón de herencia autosómico recesivo. La neutropenia es de moderada a grave, con una tasa de mortalidad del 15-25%, y el síndrome se presenta en la infancia, con infecciones recurrentes, diarrea y dificultad para alimentarse. Puede producirse enanismo, condrodisplasia e insuficiencia pancreática exocrina.
El síndrome de Shwachman-Diamond y la disqueratosis congénita (DC) ligada al cromosoma X, la hipoplasia cartilaginosa (CHH) y la anemia de Diamond-Blackfan (DBA) parecen compartir defectos genéticos comunes relacionados con la síntesis de ribosomas. La mayoría de los casos de síndrome de Shwachman-Diamond están causados por mutaciones en el gen SBDS. La función precisa de este gen aún se está dilucidando; sin embargo, está implicado en la síntesis de ribosomas y en las reacciones de procesamiento del ARN. El tratamiento es G-CSF.
En la CHH, el patrón de herencia es autosómico recesivo en el cromosoma 9, y se observa en familias amish y finlandesas. La CHH está causada por mutaciones en el gen RMRP, que codifica el componente de ARN del complejo de procesamiento del ARN mitocondrial (RNasa MRP). La neutropenia es de moderada a grave. La CHH se presenta con defectos de inmunidad celular, anemia macrocítica, enfermedad gastrointestinal y enanismo. También muestra una predisposición al cáncer, especialmente al linfoma. El tratamiento es el trasplante de médula ósea.
La disqueratosis congénita (síndrome de Zinsser-Cole-Engman) se presenta con retraso mental, pancitopenia e inmunidad celular defectuosa. La disqueratosis congénita es más común en hombres que en mujeres y es hematológicamente similar a la anemia de Fanconi. La disqueratosis congénita suele ser recesiva ligada al cromosoma X, aunque existen formas autosómicas dominantes y autosómicas recesivas de este trastorno.
La forma recesiva ligada al cromosoma X del trastorno se ha relacionado con mutaciones en DKC1, que codifica la disquerina, una proteína nucleolar asociada a las partículas de ribonucleoproteína. La forma autosómica dominante se asocia a mutaciones en otro gen, el TERC, que forma parte de la telomerasa. La telomerasa tiene un componente de proteína y otro de ARN, y el TERC codifica el componente de ARN. Los pacientes con este trastorno tienen telómeros más cortos de lo normal. El tratamiento es G-CSF, factor estimulante de colonias de granulocitos-macrófagos (GM-CSF) y trasplante de médula ósea.
El síndrome de Barth es un trastorno recesivo ligado al cromosoma X que se presenta con cardiomiopatía en la infancia, miopatía esquelética, infecciones recurrentes, enanismo y neutropenia de moderada a grave.
El síndrome de Chediak-Higashi es un trastorno autosómico recesivo con infecciones recurrentes, retraso mental, fotofobia, nistagmo, albinismo oculocutáneo, neuropatía, trastornos hemorrágicos, gingivitis y gránulos lisosomales en varias células. La neutropenia es de moderada a grave, y el tratamiento es el trasplante de médula ósea.
Mielocatexis
La mielocatexis se presenta en la infancia con neutropenia moderada y se asocia a infecciones recurrentes. La afección se debe a la apoptosis acelerada y a la disminución de la expresión de bcl-x en los precursores de neutrófilos. Se observa un aspecto nuclear anormal, con hipersegmentación con filamentos nucleares, picnosis y vacuolización citoplasmática. El tratamiento es G-CSF y GM-CSF.
Síndrome de leucocitos perezosos
El síndrome de leucocitos perezosos es una neutropenia grave con motilidad anormal de los neutrófilos asociada. La etiología es desconocida y el tratamiento es de apoyo.
Trastornos metabólicos
Son neutropenias crónicas con CNA variables. They include glycogen storage disease type 1b and various acidemias, such as isovaleric, propionic, and methylmalonic. In glycogen storage disease type 1b, the treatment is G-CSF and GM-CSF.
Acquired neutropenia caused by intrinsic bone marrow disease
Intrinsic bone marrow diseases that may cause neutropenia include the following:
-
Aplastic anemia
-
Hematologic malignancy (eg, leukemia, lymphoma, myelodysplasia, myeloma)
-
Ionizing radiation
-
Tumor infiltration
-
Granulomatous infection
-
Myelofibrosis
Immune-mediated neutropenia
A drug may act as a hapten and induce antibody formation. This mechanism operates in cases due to gold, aminopyrine, and antithyroid drugs. The antibodies destroy the granulocytes and may not require the continued presence of the drug for their action. Alternatively, the drug may form immune complexes that attach to the neutrophils. This mechanism operates with quinidine.
Drug immune-mediated neutropenia may be caused by the following:
-
Aminopyrine
-
Quinidine
-
Cephalosporins
-
Penicillins
-
Sulfonamides
-
Phenothiazines
-
Hydralazine
Other medications have been implicated
Autoimmune neutropenia is the neutrophil analogue of autoimmune hemolytic anemia and of idiopathic thrombocytopenic neutropenia. It should be considered in the absence of any of the common causes. Antineutrophil antibodies have been demonstrated in these patients. Autoimmune neutropenia may be associated with the following:
-
Rheumatoid arthritis (with or without Felty syndrome)
-
Sjögren syndrome
-
Chronic, autoimmune hepatitis
-
Systemic lupus erythematosus
-
Thymoma
-
Goodpasture disease
-
Granulomatosis with polyangiitis (Wegener granulomatosis)
-
Pure red blood cell (RBC) aplasia, in which there is complete disappearance of granulocyte tissue from the bone marrow; pure RBC dysplasia is a rare disorder due to the presence of antibody-mediated, granulocyte-macrophage colony forming unit (GM-CFU) inhibitory activity, and it is often associated with thymoma
-
Transfusion reactions, which can be caused by the surface antigens of neutrophilia; Los receptores de transfusiones repetidas de granulocitos podrían aloinmunizarse
-
Proliferación de linfocitos granulares grandes o leucemia
-
Bacterial sepsis
-
Viral infections (eg, influenza, measles, Epstein Barr virus , cytomegalovirus , viral hepatitis, human immunodeficiency virus -1) (see first image below)
-
Toxoplasmosis
-
Brucellosis
-
Typhoid
-
Tuberculosis (see second and third images below)
-
Malaria
-
Dengue fever
-
Rickettsial infection
-
Babesiosis
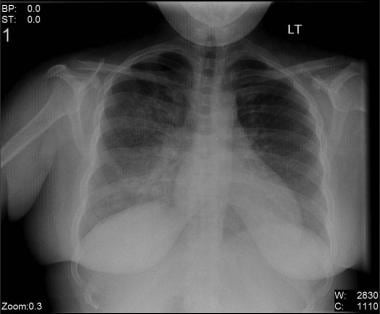 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia. 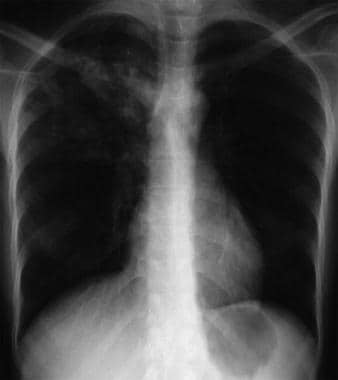 Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mujer fue admitida en aislamiento y se inició empíricamente con un régimen de 4 fármacos en el servicio de urgencias. La tuberculosis se confirmó en las pruebas de esputo. Imagen cortesía de Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mujer fue admitida en aislamiento y se inició empíricamente con un régimen de 4 fármacos en el servicio de urgencias. La tuberculosis se confirmó en las pruebas de esputo. Imagen cortesía de Remote Medicine, remotemedicine.org. 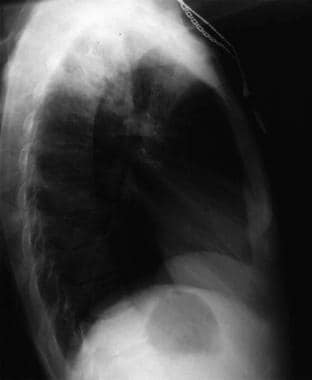 Radiografía lateral de tórax en un paciente de 31 años con neumonía por gripe. Imagen cortesía de Remote Medicine, remotemedicine.org.
Radiografía lateral de tórax en un paciente de 31 años con neumonía por gripe. Imagen cortesía de Remote Medicine, remotemedicine.org.-
Phenothiazine
-
Antithyroid drugs (thiouracil and propylthiouracil)
-
Aminopyrine
-
Chloramphenicol
-
Sulfonamides
-
Acute bacterial infection
-
Glucocorticoid administration
-
Physical stress
-
Thymoma
-
Anaphylaxis
-
Acute infection
-
Drug-induced hypersensitivity
-
Congenital absence of basophils
-
Hemorrhage
-
Hyperthyroidism
-
Ionizing radiation
-
Neoplasia
-
Ovulation
-
Urticaria
-
Drugs (eg, corticosteroid, adrenocorticotropic hormone therapy, chemotherapeutic agents, thyroid hormones)
Los organismos más comúnmente implicados son de la flora endógena. Los organismos Staphylococcus aureus se encuentran en casos de infecciones cutáneas. Los organismos gramnegativos se observan en las infecciones del tracto urinario y gastrointestinal, especialmente las especies Escherichia coli y Pseudomonas. También pueden producirse infecciones por Candida albicans. Puede encontrarse una flora mixta en la cavidad oral.
Las infecciones virales suelen provocar una neutropenia leve o moderada. La agranulocitosis es infrecuente pero puede ocurrir. Los organismos más comunes son el virus de Epstein-Barr, el virus de la hepatitis B, el virus de la fiebre amarilla, el citomegalovirus y la gripe. Muchas infecciones abrumadoras, tanto virales como bacterianas, pueden causar neutropenia grave.
Nutropenia adquirida causada por deficiencia nutricional
Las deficiencias nutricionales que pueden causar neutropenia incluyen la vitamina B-12, el folato y la deficiencia de cobre.
Nutropenia adquirida causada por fármacos y sustancias químicas, excluyendo la quimioterapia citotóxica
Se han asociado numerosos fármacos con la neutropenia. Las categorías de mayor riesgo son los medicamentos antitiroideos, los macrólidos y las procainamidas. Como ya se ha dicho, muchos fármacos actúan por un mecanismo inmunomediado. Sin embargo, algunos fármacos parecen tener efectos tóxicos directos sobre las células madre de la médula o los precursores de los neutrófilos en el compartimento mitótico. Por ejemplo, fármacos como los antipsicóticos y antidepresivos y el cloranfenicol pueden actuar como tóxicos directos en algunos individuos, basándose en el metabolismo y la sensibilidad de este modo. Otros fármacos pueden tener una combinación de mecanismos inmunes y no inmunes o pueden tener mecanismos de acción desconocidos.
Los antimicrobianos incluyen la penicilina, las cefalosporinas, la vancomicina, el cloranfenicol, la gentamicina, la clindamicina, la doxiciclina, la flucitosina, la nitrofurantoína, la novobiocina, la minociclina, la griseofulvina, la lincomicina, el metronidazol, la rifampicina, la isoniazida, la estreptomicina, tiacetazona, mebendazol, pirimetamina, levamisol, ristocetina, sulfonamidas, cloroquina, hidroxicloroquina, quinacrina, etambutol, dapsona, ciprofloxacina, trimetoprim, imipenem/cilastatina, zidovudina, fludarabina, aciclovir y terbinafina.
Los analgésicos y antiinflamatorios incluyen aminopirina, dipirona, indometacina, ibuprofeno, ácido acetilsalicílico, diflunisal, sulindac, tolmetina, benoxaprofeno, barbitúricos, mesalazina y quinina.
Los antipsicóticos, antidepresivos y agentes neurofarmacológicos incluyen fenotiazinas (clorpromazina, metilpromazina, mepazina, promazina, tioridazina, proclorperazina, trifluoperazina, trimeprazina), clozapina, risperidona, imipramina, desipramina, diazepam, clordiazepóxido, amoxapina, meprobamato, tiotixeno y haloperidol.
Los anticonvulsivos incluyen el ácido valproico, la fenitoína, la trimetadiona, la mefenitoína (Mesantoína), la etosuximida y la carbamazepina.
Los medicamentos antitiroideos incluyen el tiouracilo, el propiltiouracilo, el metimazol, el carbimazol, el perclorato de potasio y el tiocianato.
Los medicamentos cardiovasculares incluyen procainamida, captopril, aprindina, propranolol, hidralazina, metildopa, quinidina, diazóxido, nifedipina, propafenona, ticlopidina y vesnarinona.
Los antihistamínicos incluyen cimetidina, ranitidina, tripelennamina (piribenzamina), metafenileno, thenalidina, bromfeniramina y mianserina.
Los diuréticos incluyen acetazolamida, bumetanida, clorotiazida, hidroclorotiazida, clortalidona, metazolamida y espironolactona.
Los agentes hipoglucemiantes incluyen clorpropamida y tolbutamida.
Los fármacos antipalúdicos incluyen amodiaquina, dapsona, hidroxicloroquina, pirimetamina y quinina.
Los medicamentos diversos incluyen alopurinol, colchicina, aminoglutetimida, famotidina, bezafibrato, flutamida, tamoxifeno, penicilamina, ácido retinoico, metoclopramida, fenindiona, dinitrofenol, ácido etacrínico, diclorodifeniltricloroetano (DDT), cinofeno, antimonio, piritildiona, rauwolfia, etanol, clorpropamida, tolbutamida, tiazidas, espironolactona, metazolamida, acetazolamida, IGIV y levodopa.
Los metales pesados incluyen el oro, el arsénico y el mercurio.
La exposición a fármacos o productos químicos es la causa más común de agranulocitosis: aproximadamente la mitad de los pacientes tienen antecedentes de exposición a medicamentos o productos químicos. Cualquier sustancia química o fármaco que pueda deprimir la médula ósea y causar hipoplasia o aplasia es capaz de causar agranulocitosis. Algunos fármacos hacen esto a todo el mundo si se administran en dosis suficientemente grandes. Otros agentes parecen causar reacciones idiosincrásicas que afectan sólo a ciertos individuos susceptibles.
Algunos agentes (por ejemplo, el ácido valproico, la carbamazepina y los antibióticos betalactámicos) actúan por inhibición directa de la mielopoyesis. En los cultivos de médula ósea, estos agentes inhiben la formación de colonias de granulocitos de forma relacionada con la dosis. El daño directo al microambiente de la médula ósea o a los precursores mieloides desempeña un papel en la mayoría de los demás casos.
Muchos fármacos asociados a la agranulocitosis han sido notificados a la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) en virtud de su requisito de notificación de reacciones adversas. Muchos agentes también se notifican a un registro mantenido por la Asociación Médica Americana (AMA). Los fármacos notificados se utilizaron solos, en combinación con otro fármaco conocido como potencialmente tóxico, o con otro fármaco sin toxicidad conocida. Varios fármacos son especialmente destacados por su alta frecuencia de asociación con la agranulocitosis. Entre ellos se encuentran los siguientes:
Miscellaneous immunologic neutropenias
Immunologic neutropenias may occur after bone marrow transplantation and blood product transfusions.
Felty syndrome is a syndrome of rheumatoid arthritis, splenomegaly, and neutropenia. Splenectomy shows an initial response, but neutropenia may recur in 10-20% of patients. Treatment is directed toward rheumatoid arthritis.
In complement activation–mediated neutropenia, hemodialysis, cardiopulmonary bypass, and extracorporeal membrane oxygenation (ECMO) expose blood to artificial membranes and can cause complement activation with subsequent neutropenia.
In splenic sequestration, the degree of neutropenia resulting from this process is proportional to the severity of the splenomegaly and the bone marrow’s ability to compensate for the reduction in circulating bands and neutrophils.
Eosinopenia and basophilopenia
Eosinopenia may be associated with the following:
Decreased circulating basophils may be associated with the following:
Go to Pediatric Autoimmune and Chronic Benign Neutropenia for complete information on this topic.
-
En la neutropenia neonatal isoinmune, la madre produce anticuerpos antineutrófilos IgG contra antígenos neutrófilos fetales que se reconocen como no propios. Esto ocurre en el 3% de los nacidos vivos. El trastorno se manifiesta como fiebre neonatal, infección del tracto urinario, celulitis, neumonía y sepsis. La duración de la neutropenia suele ser de 7 semanas.
La neutropenia crónica autoinmune se observa en adultos y no tiene predilección por la edad. Hasta un 36% de los pacientes presentarán anticuerpos antineutrófilos en suero, y el curso clínico suele ser menos grave. Los pacientes pueden presentar este trastorno en asociación con el lupus eritematoso sistémico, la artritis reumatoide, la granulomatosis de Wegener y la hepatitis crónica.
Si la neutropenia autoinmune crónica está asociada a estas enfermedades, los corticosteroides están indicados como tratamiento. En neonatos y niños, este trastorno se asocia a un menor riesgo de infección y a infecciones más leves que afectan al oído medio, al tracto gastrointestinal y a la piel.
La linfocitosis T-gamma, o trastorno linfoproliferativo, es una enfermedad clonal de linfocitos T CD3+ o células asesinas naturales (NK) CD3- que se infiltran en la médula ósea y en los tejidos. También conocida como leucemia de linfocitos granulares grandes (LGL-leucemia), la linfocitosis T-gamma puede estar asociada a la artritis reumatoide y a los anticuerpos antineutrófilos de título elevado. La neutropenia es persistente y grave. El tratamiento suele ser de apoyo, pero también está dirigido a eliminar la población clonal.
Neutropenia adquirida causada por infección
Las infecciones son la forma más común de neutropenia adquirida. Las infecciones que pueden causar neutropenia incluyen, entre otras, las siguientes:
 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.  Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mujer fue admitida en aislamiento y se inició empíricamente con un régimen de 4 fármacos en el servicio de urgencias. La tuberculosis se confirmó en las pruebas de esputo. Imagen cortesía de Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mujer fue admitida en aislamiento y se inició empíricamente con un régimen de 4 fármacos en el servicio de urgencias. La tuberculosis se confirmó en las pruebas de esputo. Imagen cortesía de Remote Medicine, remotemedicine.org. 