Neutropénie
La liste pour toutes les causes potentielles de neutropénie n’est pas courte. L’étiologie de la neutropénie peut conceptuellement être envisagée de deux grandes façons, par mécanisme ou par catégorie étiologique.
Les mécanismes à l’origine de la neutropénie sont variés et pas complètement compris. Dans de nombreux cas, la neutropénie survient après une exposition prolongée à un médicament ou à une autre substance, entraînant une diminution de la production de neutrophiles par une moelle osseuse hypoplasique. Cela suggère un effet toxique direct sur les cellules souches. Dans d’autres cas, une exposition répétée mais intermittente à un médicament ou à une autre substance est nécessaire. Cela suggère un mécanisme immunitaire, bien que cette idée n’ait pas été prouvée. Dans de nombreuses situations cliniques, l’exposition exacte et sa durée par rapport à l’apparition de la neutropénie ne sont pas connues.
Au vu de cette compréhension incomplète des mécanismes de la neutropénie, une classification par grande catégorie étiologique est plus simple à retenir. Dans ce schéma, l’étiologie de la neutropénie peut être classée comme congénitale (héréditaire) ou acquise. Bien que cette catégorisation puisse avoir une utilité limitée pour le diagnostic clinique, elle peut être utile pour séparer clairement les causes héréditaires de la neutropénie de la panoplie des causes acquises. Dans le cadre des neutropénies héréditaires, ces troubles peuvent être décrits plus précisément comme étant associés à une neutropénie isolée ou à d’autres défauts, qu’ils soient immunitaires ou phénotypiques.
De nombreux troubles héréditaires sont dus à des mutations du gène codant pour l’élastase des neutrophiles, ELA2. Plusieurs allèles sont impliqués. Les mutations les plus fréquentes sont des substitutions introniques qui inactivent un site d’épissage dans l’intron 4. Des gènes autres que ELA2 sont également impliqués. Le tableau ci-dessous énumère certaines des conditions génétiques impliquées ; ce sont des conditions peu courantes.
Tableau 1. Conditions génétiques (héréditaires) dans l’agranulocytose (Ouvrir le tableau dans une nouvelle fenêtre)
|
Syndrome |
Inheritance |
Gene |
Clinical Features |
|
Cyclic neutropenia |
Autosomal dominant |
ELA2 |
Alternate 21-day cycling of neutrophils and monocytes |
|
Kostmann syndrome |
Autosomal recessive |
Unknown |
Stable neutropenia, no MDS or AML |
|
Severe congenital neutropenia |
Autosomal dominant |
ELA2 (35-84%) |
Stable neutropenia, MDS or AML |
|
Autosomal dominant |
GFI1 |
Stable neutropenia, circulating myeloid progenitors, lymphopenia |
|
|
Sex linked |
Wasp |
Neutropenic variant of Wiskott-Aldrich syndrome |
|
|
Autosomal dominant |
G-CSFR |
G-CSF–refractory neutropenia, no AML or MDS |
|
|
Hermansky-Pudlak syndrome type 2 |
Autosomal recessive |
AP3B1 |
Severe congenital neutropenia, platelet dense-body defect, oculocutaneous albinism |
|
Chediak-Higashi syndrome |
Autosomal recessive |
LYST |
Neutropenia, oculocutaneous albinism, giant lysosomes, impaired platelet function |
|
Barth syndrome |
Sex linked |
TAZ |
Neutropenia, often cyclic; cardiomyopathy, methylglutaconic aciduria |
|
Cohen syndrome |
Autosomal recessive |
COH1 |
Neutropenia, mental retardation, dysmorphism |
|
Source: Modified from Berliner et al, 2004. AML = acute myeloid leukemia; G-CSF = granulocyte colony-stimulating factor; MDS = myelodysplastic syndrome. |
|||
Les causes de la neutropénie acquise sont complexes, mais la plupart sont liées à trois grandes catégories : l’infection, les médicaments (toxiques directs ou à médiation immunitaire) et l’auto-immunité. La neutropénie chronique bénigne, ou neutropénie chronique idiopathique, semble être un trouble chevauchant les formes héréditaires et acquises, et est parfois indiscernable. Certains patients neutropéniques présentent une histoire et un modèle familial clairs, tandis que d’autres n’ont pas d’histoire familiale, peu de déterminations d’analyses sanguines et une durée de neutropénie inconnue. Ce groupe de patients pourrait présenter une neutropénie héréditaire ou acquise. Voici un bref résumé des troubles neutropéniques congénitaux et acquis.
Nutropénie congénitale avec anomalies immunitaires associées
La neutropénie avec immunoglobulines anormales est observée chez les personnes atteintes d’agammaglobulinémie liée à l’X, de déficit isolé en immunoglobuline A (IgA), de syndrome d’hyperimmunoglobuline M liée à l’X (XHIGM) et de dysgammaglobulinémie de type I. Dans le cas du syndrome XHIGM, qui est dû à des mutations du ligand CD40, les patients peuvent en fait présenter des taux normaux ou élevés d’IgM mais des taux sériques d’IgG nettement réduits. Dans tous ces troubles, le risque d’infection est élevé, et le traitement est l’immunoglobuline intraveineuse (IVIG).
Les patients atteints de dysgénésie réticulaire présentent une neutropénie sévère, une immunité à médiation cellulaire nulle, une agammaglobulinémie et une lymphopénie. Des infections mettant en jeu le pronostic vital surviennent et sont réfractaires au facteur de stimulation des colonies de granulocytes (G-CSF). La greffe de moelle osseuse est le traitement de choix.
Nutropénies congénitales ou chroniques
La neutropénie congénitale sévère (NSC), ou syndrome de Kostmann, est le plus souvent causée par une transmission récessive et se retrouve dans des populations éloignées et isolées avec un haut degré de consanguinité. Des cas autosomiques dominants et sporadiques ont également été rapportés, le plus souvent dus à des mutations du récepteur du G-CSF. Il n’existe pas de défaut génétique uniforme dans ce syndrome. Les mutations dans ELA2, qui sont à l’origine de la neutropénie cyclique (voir ci-dessous) ne suffisent pas à expliquer le phénotype du SCN de type Kostmann.
Les patients se présentent vers l’âge de 3 mois avec des infections bactériennes récurrentes. La bouche et le péri rectum sont les sites d’infection les plus fréquents. Ce type de neutropénie est sévère, et le traitement est le G-CSF. Le risque de conversion en syndrome myélodysplasique (SMD)/leucémie myélogène aiguë (LMA) avec monosomie 7 après des traitements par G-CSF est associé à des mutations supplémentaires acquises. La plupart de ces cas sont causés par une mutation du récepteur du G-CSF. Les patients dont l’état répond cliniquement au G-CSF sont traités à vie.
Certains patients présentant d’autres formes de NSC semblent avoir des mutations dans GFI1, un gène répresseur transcriptionnel à doigts de zinc impliqué dans la fonction des cellules souches hématopoïétiques et les décisions d’engagement de lignage.
La neutropénie cyclique (NC) se caractérise par des accès périodiques de neutropénie associés à une infection, suivis d’une récupération du nombre de neutrophiles périphériques. Sa périodicité est d’environ 21 jours (fourchette, 12-35 j). Les précurseurs granulocytaires disparaissent de la moelle avant chaque nadir de neutrophiles du cycle en raison de l’apoptose accélérée des cellules progénitrices myéloïdes. Certains cas peuvent être déterminés génétiquement avec une transmission autosomique récessive. D’autres cas peuvent être dus à une transmission autosomique dominante. Dans certains cas sporadiques de CN, les patients présentent des mutations dans ELA2.
Les personnes atteintes de CN se présentent généralement comme des nourrissons ou des enfants, mais il existe des formes acquises de CN à l’âge adulte. Le pronostic est bon, avec une évolution bénigne ; cependant, 10 % des patients connaîtront des infections potentiellement mortelles. Le traitement de la neutropénie cyclique est le G-CSF quotidien.
Nutropénie chronique bénigne
La neutropénie chronique bénigne familiale, ou neutropénie ethnique bénigne, est un trouble à transmission autosomique dominante observé dans les populations d’ascendance africaine, juive yéménite, juive éthiopienne, arabe, caribéenne et antillaise. Dans les populations d’ascendance africaine et juive yéménite, les études génétiques montrent une forte association avec un polymorphisme mononucléotidique dans le gène DARC. Les patients sont généralement asymptomatiques et les infections sont bénignes. Les personnes atteintes de neutropénie chronique bénigne ont un risque d’infection globalement faible et aucune thérapie spécifique n’est nécessaire.
Dans les neutropénies chroniques bénignes non familiales, des infections légères à évolution bénigne caractérisent ce trouble. La CNA, cependant, réagit au stress, tel que l’infection, les corticostéroïdes et les catécholamines.
Nutropénie chronique sévère idiopathique
La neutropénie chronique sévère idiopathique est un diagnostic d’exclusion. Les patients atteints présentent des infections et une neutropénie sévère.
Neutropénie associée à des anomalies phénotypiques
Le syndrome de Shwachman (Shwachman-Diamond) a un mode de transmission autosomique récessif. La neutropénie est modérée à sévère, avec un taux de mortalité de 15-25%, et le syndrome se présente dans la petite enfance, avec des infections récurrentes, de la diarrhée, et des difficultés à s’alimenter. Un nanisme, une chondrodysplasie et une insuffisance pancréatique exocrine peuvent survenir.
Le syndrome de Shwachman-Diamond et la dyskératose congénitale (DC) liée à l’X, l’hypoplasie des poils de cartilage (CHH) et l’anémie de Diamond-Blackfan (DBA) semblent tous partager des défauts génétiques communs impliqués dans la synthèse des ribosomes. La plupart des cas de syndrome de Shwachman-Diamond sont causés par des mutations du gène SBDS. La fonction précise de ce gène est encore en cours d’élucidation ; toutefois, il est impliqué dans la synthèse des ribosomes et les réactions de traitement de l’ARN. Le traitement est le G-CSF.
Dans le cas du CHH, le mode de transmission est autosomique récessif sur le chromosome 9, et il est observé dans les familles amish et finlandaises. La CHH est causée par des mutations du gène RMRP, qui code pour le composant ARN du complexe ribonucléase de traitement de l’ARN mitochondrial (RNase MRP). La neutropénie est modérée à sévère. Le CHH présente des défauts de l’immunité à médiation cellulaire, une anémie macrocytaire, une maladie gastro-intestinale et un nanisme. Elle présente également une prédisposition au cancer, notamment au lymphome. Le traitement consiste en une greffe de moelle osseuse.
La dyskératose congénitale (syndrome de Zinsser-Cole-Engman) présente un retard mental, une pancytopénie et un défaut d’immunité à médiation cellulaire. La dyskératose congénitale est plus fréquente chez les hommes que chez les femmes et est hématologiquement similaire à l’anémie de Fanconi. La dyskératose congénitale est généralement récessive liée au chromosome X, bien qu’il existe des formes autosomiques dominantes et autosomiques récessives de cette maladie.
La forme récessive liée au chromosome X de la maladie a été liée à des mutations dans DKC1, qui code pour la dyskérine, une protéine nucléolaire associée aux particules de ribonucléoprotéines. La forme autosomique dominante est associée à des mutations dans un autre gène, TERC, qui fait partie de la télomérase. La télomérase a une composante protéique et une composante ARN, et TERC code la composante ARN. Les patients atteints de cette maladie ont des télomères plus courts que la normale. Le traitement est le G-CSF, le facteur de stimulation des colonies de granulocytes-macrophages (GM-CSF) et la greffe de moelle osseuse.
Le syndrome de Barth est une maladie récessive liée au chromosome X présentant une cardiomyopathie dans la petite enfance, une myopathie squelettique, des infections récurrentes, un nanisme et une neutropénie modérée à sévère.
Le syndrome de Chediak-Higashi est une maladie autosomique récessive présentant des infections récurrentes, un ralentissement mental, une photophobie, un nystagmus, un albinisme oculo-cutané, une neuropathie, des troubles de la coagulation, une gingivite et des granules lysosomaux dans diverses cellules. La neutropénie est modérée à sévère et le traitement est la greffe de moelle osseuse.
Myelokathexis
La myelokathexis se présente dans la petite enfance avec une neutropénie modérée et est associée à des infections récurrentes. Cette affection est due à une apoptose accélérée et à une diminution de l’expression de bcl-x dans les précurseurs des neutrophiles. Un aspect nucléaire anormal est observé, avec une hypersegmentation avec des brins nucléaires, une pyknose et une vacuolisation cytoplasmique. Le traitement repose sur le G-CSF et le GM-CSF.
Syndrome des leucocytes paresseux
Le syndrome des leucocytes paresseux est une neutropénie sévère associée à une motilité anormale des neutrophiles. L’étiologie est inconnue et le traitement est de nature supportive.
Les troubles métaboliques
Il s’agit de neutropénies chroniques avec des ANC variables. They include glycogen storage disease type 1b and various acidemias, such as isovaleric, propionic, and methylmalonic. In glycogen storage disease type 1b, the treatment is G-CSF and GM-CSF.
Acquired neutropenia caused by intrinsic bone marrow disease
Intrinsic bone marrow diseases that may cause neutropenia include the following:
-
Aplastic anemia
-
Hematologic malignancy (eg, leukemia, lymphoma, myelodysplasia, myeloma)
-
Ionizing radiation
-
Tumor infiltration
-
Granulomatous infection
-
Myelofibrosis
Immune-mediated neutropenia
A drug may act as a hapten and induce antibody formation. This mechanism operates in cases due to gold, aminopyrine, and antithyroid drugs. The antibodies destroy the granulocytes and may not require the continued presence of the drug for their action. Alternatively, the drug may form immune complexes that attach to the neutrophils. This mechanism operates with quinidine.
Drug immune-mediated neutropenia may be caused by the following:
-
Aminopyrine
-
Quinidine
-
Cephalosporins
-
Penicillins
-
Sulfonamides
-
Phenothiazines
-
Hydralazine
Other medications have been implicated
Autoimmune neutropenia is the neutrophil analogue of autoimmune hemolytic anemia and of idiopathic thrombocytopenic neutropenia. It should be considered in the absence of any of the common causes. Antineutrophil antibodies have been demonstrated in these patients. Autoimmune neutropenia may be associated with the following:
-
Rheumatoid arthritis (with or without Felty syndrome)
-
Sjögren syndrome
-
Chronic, autoimmune hepatitis
-
Systemic lupus erythematosus
-
Thymoma
-
Goodpasture disease
-
Granulomatosis with polyangiitis (Wegener granulomatosis)
-
Pure red blood cell (RBC) aplasia, in which there is complete disappearance of granulocyte tissue from the bone marrow; pure RBC dysplasia is a rare disorder due to the presence of antibody-mediated, granulocyte-macrophage colony forming unit (GM-CFU) inhibitory activity, and it is often associated with thymoma
-
Transfusion reactions, which can be caused by the surface antigens of neutrophilia; les receveurs de transfusions répétées de granulocytes pourraient devenir allo-immunisés
-
Prolifération des lymphocytes à gros grains ou leucémie
Dans la neutropénie néonatale isoimmune, la mère produit des anticorps antineutrophiles IgG contre des antigènes neutrophiles fœtaux reconnus comme non-soi. Ce phénomène se produit dans 3 % des naissances vivantes. Ce trouble se manifeste par une fièvre néonatale, une infection des voies urinaires, une cellulite, une pneumonie et une septicémie. La durée de la neutropénie est généralement de 7 semaines.
La neutropénie chronique auto-immune est observée chez les adultes et n’a pas de prédilection pour l’âge. Jusqu’à 36 % des patients présentent des anticorps antineutrophiles sériques et l’évolution clinique est généralement moins sévère. Les patients peuvent présenter ce trouble en association avec un lupus érythémateux systémique, une polyarthrite rhumatoïde, une granulomatose de Wegener et une hépatite chronique.
Si la neutropénie chronique auto-immune est associée à ces maladies, les corticostéroïdes sont indiqués comme traitement. Chez les nouveau-nés et les enfants, ce trouble est associé à un risque moindre d’infection et à des infections plus légères touchant l’oreille moyenne, le tube digestif et la peau.
La lymphocytose T-gamma, ou trouble lymphoprolifératif, est une maladie clonale des lymphocytes T CD3+ ou des cellules tueuses naturelles (NK) CD3- qui infiltrent la moelle osseuse et les tissus. Également connue sous le nom de leucémie des grands lymphocytes granulaires (LGL-leucémie), la lymphocytose T-gamma peut être associée à la polyarthrite rhumatoïde et est associée à des anticorps antineutrophiles de titre élevé. La neutropénie est persistante et sévère. Le traitement est souvent de nature supportive, mais il vise également à éliminer la population clonale.
Nutropénie acquise causée par une infection
Les infections sont la forme la plus courante de neutropénie acquise. Les infections qui peuvent provoquer une neutropénie comprennent, sans s’y limiter, les éléments suivants :
-
Bacterial sepsis
-
Viral infections (eg, influenza, measles, Epstein Barr virus , cytomegalovirus , viral hepatitis, human immunodeficiency virus -1) (see first image below)
-
Toxoplasmosis
-
Brucellosis
-
Typhoid
-
Tuberculosis (see second and third images below)
-
Malaria
-
Dengue fever
-
Rickettsial infection
-
Babesiosis
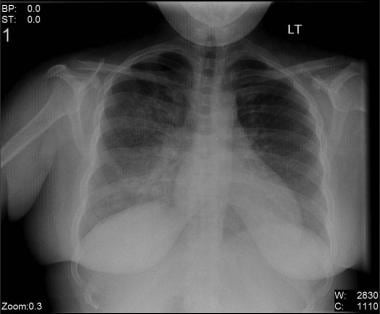 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia. 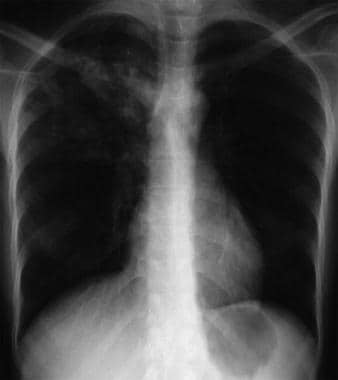 Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Cette femme a été admise en isolement et a reçu un traitement empirique de 4 médicaments aux urgences. La tuberculose a été confirmée par des tests d’expectoration. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Cette femme a été admise en isolement et a reçu un traitement empirique de 4 médicaments aux urgences. La tuberculose a été confirmée par des tests d’expectoration. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org. 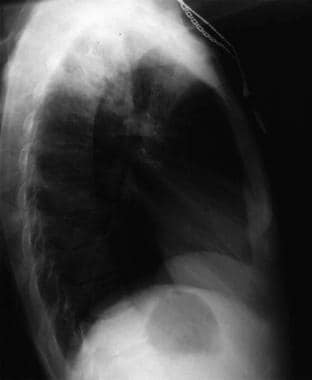 Radiographie latérale du thorax chez un patient de 31 ans atteint de pneumonie grippale. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org.
Radiographie latérale du thorax chez un patient de 31 ans atteint de pneumonie grippale. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org.
 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.  Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Cette femme a été admise en isolement et a reçu un traitement empirique de 4 médicaments aux urgences. La tuberculose a été confirmée par des tests d’expectoration. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Cette femme a été admise en isolement et a reçu un traitement empirique de 4 médicaments aux urgences. La tuberculose a été confirmée par des tests d’expectoration. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org.  Radiographie latérale du thorax chez un patient de 31 ans atteint de pneumonie grippale. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org.
Radiographie latérale du thorax chez un patient de 31 ans atteint de pneumonie grippale. Image reproduite avec l’aimable autorisation de Remote Medicine, remotemedicine.org. Les organismes les plus fréquemment impliqués proviennent de la flore endogène. Les organismes de type Staphylococcus aureus sont retrouvés dans les cas d’infections cutanées. Les organismes à Gram négatif sont observés dans les infections des voies urinaires et gastro-intestinales, notamment les espèces Escherichia coli et Pseudomonas. Des infections à Candida albicans peuvent également survenir. Une flore mixte peut être trouvée dans la cavité buccale.
Les infections virales entraînent souvent une neutropénie légère ou modérée. L’agranulocytose est peu fréquente mais peut se produire. Les organismes les plus courants sont le virus d’Epstein-Barr, le virus de l’hépatite B, le virus de la fièvre jaune, le cytomégalovirus et la grippe. De nombreuses infections écrasantes, virales et bactériennes, peuvent provoquer une neutropénie sévère.
Nutropénie acquise causée par une carence nutritionnelle
Les carences nutritionnelles qui peuvent provoquer une neutropénie comprennent la vitamine B-12, les folates et la carence en cuivre.
Nutropénie acquise causée par des médicaments et des produits chimiques, à l’exception de la chimiothérapie cytotoxique
De nombreux médicaments ont été associés à la neutropénie. Les catégories les plus à risque sont les médicaments antithyroïdiens, les macrolides et les procaïnamides. Comme indiqué ci-dessus, de nombreux médicaments agissent par un mécanisme à médiation immunitaire. Cependant, certains médicaments semblent avoir des effets toxiques directs sur les cellules souches de la moelle ou les précurseurs des neutrophiles dans le compartiment mitotique. Par exemple, des médicaments tels que les antipsychotiques et antidépresseurs et le chloramphénicol peuvent agir comme des toxines directes chez certains individus, en fonction de leur métabolisme et de leur sensibilité à cet égard. D’autres médicaments peuvent avoir une combinaison de mécanismes immunitaires et non immunitaires ou peuvent avoir des mécanismes d’action inconnus.
Les antimicrobiens comprennent la pénicilline, les céphalosporines, la vancomycine, le chloramphénicol, la gentamicine, la clindamycine, la doxycycline, la flucytosine, la nitrofurantoïne, la novobiocine, la minocycline, la griséofulvine, la lincomycine, le métronidazole, la rifampicine, l’isoniazide, la streptomycine, thiacétazone, mébendazole, pyriméthamine, lévamisole, ristocétine, sulfamides, chloroquine, hydroxychloroquine, quinacrine, éthambutol, dapsone, ciprofloxacine, triméthoprime, imipénème/cilastatine, zidovudine, fludarabine, acyclovir et terbinafine.
Les analgésiques et les anti-inflammatoires comprennent l’aminopyrine, la dipyrone, l’indométhacine, l’ibuprofène, l’acide acétylsalicylique, le diflunisal, le sulindac, la tolmétine, le benoxaprofène, les barbituriques, la mésalazine et la quinine.
Les antipsychotiques, les antidépresseurs et les agents neuropharmacologiques comprennent les phénothiazines (chlorpromazine, méthylpromazine, mépazine, promazine, thioridazine, prochlorpérazine, trifluopérazine, triméprazine), la clozapine, la rispéridone, l’imipramine, la désipramine, le diazépam, le chlordiazépoxide, l’amoxapine, le méprobamate, le thiothixène et l’halopéridol.
Les anticonvulsivants comprennent l’acide valproïque, la phénytoïne, la triméthadione, la méphénytoïne (Mésantoïne), l’éthosuximide et la carbamazépine.
Les antithyroïdiens comprennent le thiouracile, le propylthiouracile, le méthimazole, le carbimazole, le perchlorate de potassium et le thiocyanate.
Les médicaments cardiovasculaires comprennent la procaïnamide, le captopril, l’aprindine, le propranolol, l’hydralazine, la méthyldopa, la quinidine, le diazoxide, la nifédipine, la propafénone, la ticlopidine et la vesnarinone.
Les antihistaminiques comprennent la cimétidine, la ranitidine, la tripélennamine (Pyribenzamine), le méthaphénilène, la thénalidine, la bromphéniramine et la miansérine.
Les diurétiques comprennent l’acétazolamide, le bumétanide, le chlorothiazide, l’hydrochlorothiazide, la chlorthalidone, le méthazolamide et la spironolactone.
Les hypoglycémiants comprennent le chlorpropamide et le tolbutamide.
Les antipaludéens comprennent l’amodiaquine, la dapsone, l’hydroxychloroquine, la pyriméthamine et la quinine.
Les médicaments divers comprennent l’allopurinol, la colchicine, l’aminoglutéthimide, la famotidine, le bézafibrate, le flutamide, le tamoxifène, la pénicillamine, l’acide rétinoïque, le métoclopramide, la phénindione, le dinitrophénol, l’acide éthacrynique, dichlorodiphényltrichloroéthane (DDT), cinchophène, antimoine, pyrithyldione, rauwolfia, éthanol, chlorpropamide, tolbutamide, thiazides, spironolactone, méthazolamide, acétazolamide, IVIG et lévodopa.
Les métaux lourds comprennent l’or, l’arsenic et le mercure.
L’exposition à des médicaments ou à des produits chimiques est la cause la plus fréquente d’agranulocytose : environ la moitié des patients ont des antécédents de médicaments ou d’exposition à des produits chimiques. Tout produit chimique ou médicament qui peut déprimer la moelle osseuse et provoquer une hypoplasie ou une aplasie est capable de provoquer une agranulocytose. Certains médicaments le font pour tout le monde s’ils sont administrés à des doses suffisamment importantes. D’autres agents semblent provoquer des réactions idiosyncrasiques qui n’affectent que certains individus sensibles.
Certains agents (par exemple, l’acide valproïque, la carbamazépine et les antibiotiques bêta-lactamines) agissent par inhibition directe de la myélopoïèse. Dans les cultures de moelle osseuse, ces agents inhibent la formation de colonies de granulocytes de façon proportionnelle à la dose. Une atteinte directe du microenvironnement de la moelle osseuse ou des précurseurs myéloïdes joue un rôle dans la plupart des autres cas.
De nombreux médicaments associés à l’agranulocytose ont été signalés à la Food and Drug Administration (FDA) américaine dans le cadre de son obligation de déclaration des effets indésirables. De nombreux agents sont également signalés à un registre tenu par l’American Medical Association (AMA). Les médicaments signalés ont été utilisés seuls, en association avec un autre médicament connu pour être potentiellement toxique, ou avec un autre médicament sans toxicité connue. Plusieurs médicaments sont particulièrement importants en raison de leur fréquence élevée d’association avec l’agranulocytose. Il s’agit notamment des suivants :
-
Phenothiazine
-
Antithyroid drugs (thiouracil and propylthiouracil)
-
Aminopyrine
-
Chloramphenicol
-
Sulfonamides
Miscellaneous immunologic neutropenias
Immunologic neutropenias may occur after bone marrow transplantation and blood product transfusions.
Felty syndrome is a syndrome of rheumatoid arthritis, splenomegaly, and neutropenia. Splenectomy shows an initial response, but neutropenia may recur in 10-20% of patients. Treatment is directed toward rheumatoid arthritis.
In complement activation–mediated neutropenia, hemodialysis, cardiopulmonary bypass, and extracorporeal membrane oxygenation (ECMO) expose blood to artificial membranes and can cause complement activation with subsequent neutropenia.
In splenic sequestration, the degree of neutropenia resulting from this process is proportional to the severity of the splenomegaly and the bone marrow’s ability to compensate for the reduction in circulating bands and neutrophils.
Eosinopenia and basophilopenia
Eosinopenia may be associated with the following:
-
Acute bacterial infection
-
Glucocorticoid administration
-
Physical stress
-
Thymoma
Decreased circulating basophils may be associated with the following:
-
Anaphylaxis
-
Acute infection
-
Drug-induced hypersensitivity
-
Congenital absence of basophils
-
Hemorrhage
-
Hyperthyroidism
-
Ionizing radiation
-
Neoplasia
-
Ovulation
-
Urticaria
-
Drugs (eg, corticosteroid, adrenocorticotropic hormone therapy, chemotherapeutic agents, thyroid hormones)
Go to Pediatric Autoimmune and Chronic Benign Neutropenia for complete information on this topic.