Determinazione della mutazione V617F del gene JAK2 nelle sindromi mieloproliferative croniche nel nostro paese: un case report
Determinazione della mutazione V617F del gene JAK2 nelle sindromi mieloproliferative croniche nel nostro paese: un case report
I dottori Daniela Lens*, Pablo Muxi†, Lics. Andreína Brugnini‡,
Natalia Trías‡, Dra. Silvia Pierri§
Ospedale di Clínicas. Facoltà di Medicina.
Università della Repubblica. Montevideo, Uruguay
Abstract
La policitemia vera (PV), la trombocitemia essenziale (ET) e la mielofibrosi idiopatica (IM) sono disturbi mieloproliferativi clonali strettamente correlati, caratterizzati da un’eccessiva proliferazione di uno o più lignaggi mieloidi come eritrociti, piastrine e fibroblasti nel midollo osseo.
Mentre esistono criteri rigorosi per la diagnosi di queste sindromi mieloproliferative, la categorizzazione precisa rimane oggetto di dibattito e inoltre questi disturbi sono difficili da differenziare dai processi reattivi in molti casi.
Di recente, nel 2005, una mutazione nel gene della tirosin-chinasi Janus kinase 2 (JAK2) è stata identificata in diverse di queste entità. Questa mutazione comporta la sostituzione di una G con una T in posizione 1849, con conseguente sostituzione di una fenilalanina con una valina nella proteina (JAK2 V617F).
L’incidenza di questa mutazione è stata osservata in circa il 90% dei casi con PV e in circa il 50% dei casi con IM e ET.
In questo articolo descriveremo l’individuazione di questa mutazione in un paziente con una diagnosi di probabile PV utilizzando un test di reazione a catena della polimerasi (PCR) allele-specifico altamente sensibile per l’individuazione di questa mutazione e discuteremo l’importanza di questa mutazione recentemente scoperta nella diagnosi e nel trattamento delle sindromi mieloproliferative BCR-ABL-negative.
Parole chiave: DISORDINI MIELOPROLIFERATIVI – diagnosi.
MUTAZIONE – genetica.
PROTEIN-TYROSINE KINASE.
* Professore associato. Dipartimento di Medicina di base.
† Professore associato. Clinica di ematologia. Dipartimento Clinico di Medicina.
‡ Biochimica Lic. Assistente. Dipartimento di Medicina di base.
§ Professore associato. Clinica di ematologia. Dipartimento Clinico di Medicina.
Corrispondenza: Dr. Daniela Lens.
Dipartimento. Medicina di base. Hospital de Clínicas. Piso 15. Avda Italia s/n. Montevideo CP 11600. Montevideo, Uruguay.
Email: [email protected]
Ricevuto: 31/7/06.
Accettato: 26/2/07.
Introduzione
In contrasto con la leucemia mieloide cronica dove l’identificazione del riarrangiamento specifico BCR-ABL ha portato a una svolta nella diagnosi, nel monitoraggio e nel trattamento di questa entità, nelle sindromi mieloproliferative croniche BCR-ABL negative (PMS), come la policitemia vera (PV), la trombocitemia essenziale (ET) e la mielofibrosi idiopatica (IM), fino a poco tempo fa non erano note alterazioni genetiche specifiche.
Tra maggio e giugno 2005, cinque gruppi di ricercatori hanno descritto una nuova mutazione puntiforme nel gene della tirosin-chinasi JAK2, dovuta alla sostituzione di una guanina con una timina al nucleotide 1849 nell’esone 14, con conseguente sostituzione di una valina con una fenilalanina in posizione 617 della proteina, per cui la mutazione JAK2 è denominata V617F(1-5).
Questa mutazione è stata osservata in circa il 90% dei casi con FV quando sono utilizzati saggi ad alta sensibilità per rilevarla(6). È stata anche trovata in circa il 50% dei casi con IM e ET, mentre non è stata trovata in soggetti sani o in pazienti con eritrocitosi secondaria, quindi questa mutazione ha un valore predittivo molto alto nel distinguere la SMPC da condizioni non clonali come la policitemia secondaria.
Questa mutazione avviene in una regione altamente conservata di un dominio autoinibitorio che regola negativamente la segnalazione di JAK2. Diversi studi hanno dimostrato che questa mutazione è coinvolta nella patogenesi di queste condizioni, soprattutto a causa di un guadagno di funzione del gene JAK2 e attraverso una perdita di controllo che sono associati con l’eccessiva mieloproliferazione di questi disturbi.
In questo articolo descriveremo l’individuazione di questa mutazione in un paziente con una diagnosi di probabile PV e discuteremo l’importanza di questa nuova mutazione nella diagnosi e nel trattamento delle SMP BCR-ABL negative.
Materiale e metodo
Estrazione dell’acido desossiribonucleico (DNA) è stata eseguita da 1 ml di sangue periferico citrato, utilizzando il reagente commerciale dnazol (Life Techno-logies) e seguendo il protocollo indicato dal produttore. I campioni di DNA sono stati conservati a 4°C.
Rilevamento della mutazione Vl617F
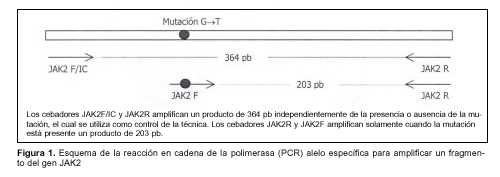
La mutazione V617F è stata rilevata dal DNA genomico precedentemente ottenuto mediante reazione a catena della polimerasi allele-specifica (PCR) secondo il metodo pubblicato da Baxter et al(1). Questa PCR è progettata per utilizzare tre diversi primer, chiamati JAK2R, JAK2F/IC e JAK2F (tabella 1). Il primer JAK2F è specifico per l’allele mutante e il primer JAK2F/IC si trova in tutti gli individui indipendentemente dalla presenza o assenza della mutazione, costituendo così un controllo interno della reazione. Così, in tutti gli individui viene amplificato un prodotto di 364 paia di basi (bp) con i primer JAK2F/IC e JAK2R, e solo negli individui con la mutazione in studio si ottiene un prodotto di 203 bp usando i primer JAK2F e JAK2R (figura 1).
L’amplificazione PCR è stata eseguita in un volume finale di 20 uL utilizzando 500 ng di DNA, 200 mM/L di ogni dNTPs, 0,5 U di Taq polimerasi (New England Biolabs) e 2 uL di 10x Taq buffer contenente 2 mM MgCl (New England Biolabs), 0,5 mM di primer JAK2 F e JAK2 F/IC e 1 mM di primer JAK2 R.
Il programma PCR utilizzato consiste in 36 cicli, iniziando con una denaturazione di 5 minuti, poi ciclando: 30 secondi a 94°C, 30 secondi a 58°C e 30 secondi a 72°C, terminando con un’estensione finale di 5 minuti. Un controllo negativo contenente tutti i reagenti necessari per l’amplificazione e dove il DNA template è sostituito da acqua è stato sempre incluso in tutte le reazioni. La verifica del prodotto di PCR atteso è stata eseguita su un gel di agarosio al 2% colorato con bromuro di etidio(7).

Analisi del caso
Paziente maschio, 72 anni, forte fumatore, con ipertensione arteriosa cronica in trattamento e portatore di una arteriopatia cronica ostruttiva che nel 2002 ha determinato un’amputazione del quinto dito del piede sinistro. Nel dicembre 2004, presentando un nuovo episodio ostruttivo all’arto inferiore sinistro, il suo emogramma ha mostrato un aumento della conta dei globuli rossi con un ematocrito del 66%, insieme a una granulocitosi di 24.000 e piastrine di 245.000. L’ematocrito è rimasto lo stesso e la conta delle piastrine è aumentata progressivamente fino a 710.000. Nello studio della biopsia del midollo osseo, è stato osservato un midollo osseo ipercellulare con iperplasia della serie eritroide, che ha portato alla conclusione di una sindrome mieloproliferativa cronica di tipo PV.
Il paziente è stato trattato con flebotomie e nel maggio 2005 ha iniziato il trattamento con idrossiurea, che è stato sospeso nel febbraio 2006 a causa di una complicazione emorragica.
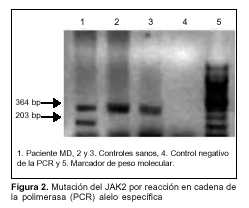
Come illustrato nella figura 2, il paziente ha mostrato un pattern a due bande nello studio PCR allele-specifico per la mutazione V617F del gene JAK2. Da un lato, la banda di controllo di 364 bp che è presente anche nei due controlli sani studiati. D’altra parte, un prodotto di 203 bp è stato amplificato anche nel paziente che mostra la presenza della mutazione. Questa banda non è stata osservata nei controlli sani.
Discussione e conclusioni
In questo lavoro abbiamo descritto il primo caso in cui la mutazione V617 del gene JAK2 è stata identificata nel nostro paese. Questo lavoro fa parte di un progetto multidisciplinare tra il Dipartimento di Medicina di Base e il Servizio di Ematologia dell’Hospital de Clínicas, al fine di determinare la presenza di questa mutazione in 100 pazienti con PMS BCR-ABL-negativa.
La recente identificazione della mutazione attivante V617F nel gene JAK2 rappresenta un importante progresso nella conoscenza della patogenesi delle sindromi mieloproliferative BCR-ABL-negative. A causa dell’alta frequenza con cui questa mutazione si trova in PV e in misura minore in ET e IM, è stato postulato che l’individuazione di questa mutazione avrebbe un forte impatto sulla diagnosi, classificazione e trattamento di queste entità. Diversi gruppi hanno proposto di incorporare l’individuazione di questa mutazione come test diagnostico di prima linea in presenza di un ematocrito superiore al 51% o quando si sospetta una PV, dato che l’individuazione di JAK2 V617F ha un valore predittivo positivo del 100% per la diagnosi di PV(8,9). D’altra parte, diversi autori hanno postulato che la presenza di questa mutazione permetterebbe una nuova classificazione della PMS, poiché ET, PV e MI che portano la mutazione JAK2 potrebbero rappresentare la stessa malattia in diversi stadi evolutivi invece di entità diverse(10-12). Tuttavia, l’esatto valore clinico e il beneficio di rilevare questa mutazione rimane da stabilire e ha bisogno di studi clinici prospettici appositamente progettati per risolvere queste domande.
Inoltre, la scoperta di questa mutazione apre nuove prospettive per terapie specificamente mirate alla proteina chinasi mutata, simili agli inibitori BCR-ABL nelle leucemie mieloidi croniche.
Sommario
La policitemia vera (PV), la trombocitemia essenziale (ET-TE) e la mielofibrosi idiopatica (IM-MI) sono disturbi mieloproliferativi clonali caratterizzati da un’eccessiva proliferazione di uno o più lignaggi mieloidi come eritrociti, piastrine e fibroblasti del midollo osseo.
La categorizzazione precisa delle sindromi mieloproliferative deve ancora essere discussa anche se i criteri diagnostici sono rigorosi; inoltre questi disturbi sono difficili da differenziare dai processi reattivi.
Di recente, nel 2005, la mutazione JAK2 è stata identificata in molte di queste entità. Il sequenziamento della regione codificante di JAK2 ha rivelato una trasversione da G a T in posizione 1849, che ha cambiato una valina in una fenilalanina (JAK2 V617F).
L’incidenza della mutazione V617F-JAK2 era quasi il 90% nei pazienti con PV, e il 50% nei pazienti con IM e ET.
In questo studio descriviamo l’individuazione della mutazione V617F-JAK2 in un paziente sospettato di PV utilizzando l’analisi allele-specifica della reazione a catena della polimerasi (PCR) e discutiamo l’importanza della mutazione per la diagnosi e il trattamento delle sindromi mieloproliferative BCR-ABL negative.
Resumé
La policitemia vera (PV), La trombocitemia essenziale (ET) e la mielofibrosi idiopatica (IM) sono disturbi mieloproliferativi clonali strettamente correlati, caratterizzati da un’eccessiva proliferazione di una o più linee mieloidi come eritrociti, piastrine e fibroblasti del midollo osseo.
Anche se ci sono criteri rigorosi per la diagnosi di queste sindromi mieloproliferative, la categorizzazione precisa rimane una questione di dibattito e, inoltre, questi disturbi sono difficili da differenziare dai processi reattivi in diverse occasioni.
Di recente, nel 2005, una mutazione nel gene della tirosin-chinasi Janus kinase 2 (JAK2) è stata identificata in diverse di queste entità. Questa mutazione consiste nella sostituzione di una G con una T in posizione 1849, con conseguente sostituzione nella proteina di una fenilalanina con la valina (JAK2 V617F).
L’incidenza di questa mutazione è stata osservata in quasi il 90% dei casi con PV e, in circa il 50% dei casi con MI e TE.
In questo lavoro descriveremo l’individuazione di questa mutazione in un paziente con una diagnosi di probabile PV per mezzo di un test di reazione a catena della polimerasi (PCR) allele-specifica con alta sensibilità per l’individuazione di questa mutazione e discuteremo l’importanza di questa mutazione appena scoperta nella diagnosi e nel trattamento di BCR-Sindromi mieloproliferative ABL negative.
Resumo
A Policitemia Vera (PV), una trombocitemia emorragica (TE) e una mielofibrosi idiopatica (MI) sono transtornos mieloproliferativos clonais fortemente correlati e caratterizzati da uma proliferação excessiva de uma ou mais línhas mielóides tais como eritrócitos, platequetas e fibroblastos da medula óssea.
Embora existam critérios rigorosos para o diagnóstico destas síndromes mieloproliferativas, a classificação precisa continua sendo discutida; ademais muitas vezes é muito difícil diferenciar estes distúrbios de processos reativos.
Nel 2005, una mutazione nel gene tirosin-chinasi Janus kinase 2 (JAK2) è stata identificata in diverse di queste entità. In questa mutazione si osserva una sostituzione da G a T in posizione 1849, che porta a una sostituzione da fenilalanina a valina (JAK2 V617F) nella proteina.
Questa mutazione è stata osservata in circa il 90% dei casi con PV e in circa il 50% dei casi con MI e TE.
In questo articolo descriviamo l’individuazione di questa mutazione in un paziente con una probabile diagnosi di PV utilizzando un test di reazione a catena della polimerasi (PCR) allele-specifico altamente sensibile per l’individuazione di questa mutazione e discutiamo l’importanza di questa nuova mutazione scoperta nella diagnosi e nel trattamento delle sindromi mieloproliferative BCR-ABL-negative.
Bibliografia
1. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Mutazione acquisita della tirosin-chinasi JAK2 nei disturbi mieloproliferativi umani. Lancet 2005; 365: 1054-61.
2. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Layout C, et al. Una mutazione clonale unica di JAK2 che porta alla segnalazione costitutiva causa la policitemia vera. Natura 2005; 434: 1144-8.
3. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. Un guadagno di mutazione funzione in JAK2 è spesso trovato in pazienti con disturbi mieloproliferativi. N Engl J Med 2005; 352: 1779-90.
4. Levine RL, Waldleigh M, Cools J, Ebert BL, Werning G, Huntly BJ, et al. Mutazione attivante nella tirosin-chinasi JAK2 nella policitemia vera, trombocitemia essenziale e metaplasia mieloide con mielofibrosi. Cancer Cell 2005; 7: 387-97.
6. Vainshenker W, Constantinescu SN. Una mutazione di attivazione unica in JAK2 (V617F) è all’origine della policitemia vera e permette una nuova classificazione delle malattie mieloproliferative. Hematology Am Soc Hematol Educ Program 2005; 195-200.
7. Sambrook J, Russell DW. Clonazione molecolare: un manuale di laboratorio. 3 ed. New York: Cold Spring Harbor Laboratory, 2001.
8. Tefferi A, Pardanani A. Screening delle mutazioni per JAK2V617F: quando ordinare il test e come interpretare i risultati. Leuk Res 2006; 30(6): 739-44.
9. James C, Delhommeau F, Marzac C, Teyssandier I, Couedic JP, Giraudier S, et al. Rilevazione di JAK2 V617F come test diagnostico di prima intenzione per l’eritrocitosi. Leucemia 2006; 20: 350-3.
10. Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, et al. Definizione di sottotipi di trombocitemia essenziale e relazione alla policitemia vera basata sullo stato di mutazione JAK2 V617F: uno studio prospettico. Lancet 2005; 366: 1945-53.
11. Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, Ponziani V, et al. Implicazioni cliniche della mutazione JAK2 V617F nella trombocitemia essenziale. Leucemia 2005; 19: 1847-9.