Neutropenia
L’elenco di tutte le potenziali cause di neutropenia non è breve. L’eziologia della neutropenia può essere concettualmente vista in due grandi modi, per meccanismo o categoria eziologica.
I meccanismi che causano neutropenia sono vari e non completamente compresi. In molti casi, la neutropenia si verifica dopo l’esposizione prolungata a un farmaco o ad altre sostanze, con conseguente diminuzione della produzione di neutrofili da parte del midollo osseo ipoplasico. Questo suggerisce un effetto tossico diretto delle cellule staminali. In altri casi, è necessaria un’esposizione ripetuta ma intermittente a un farmaco o ad altre sostanze. Questo suggerisce un meccanismo immunitario, anche se questa idea non è stata dimostrata. In molte situazioni cliniche, l’esposizione esatta e la sua durata in relazione all’insorgenza della neutropenia non sono note.
In considerazione di questa comprensione incompleta dei meccanismi della neutropenia, la classificazione per ampia categoria eziologica è più semplice da mantenere. In questo schema, l’eziologia della neutropenia può essere classificata come congenita (ereditaria) o acquisita. Anche se questa categorizzazione può avere un’utilità clinica diagnostica limitata, può essere utile per separare chiaramente le cause ereditarie di neutropenia dalla panoplia di cause acquisite. Nell’ambito delle neutropenie ereditarie, questi disturbi possono essere ulteriormente descritti come associati a neutropenia isolata o ad altri difetti, sia immunitari che fenotipici.
Molti disturbi ereditari sono dovuti a mutazioni nel gene che codifica l’elastasi neutrofila, ELA2. Sono coinvolti diversi alleli. Le mutazioni più comuni sono sostituzioni introniche che inattivano un sito di splice nell’introne 4. Sono coinvolti anche altri geni oltre a ELA2. La tabella qui sotto elenca alcune delle condizioni genetiche coinvolte; queste sono condizioni non comuni.
Tabella 1. Condizioni genetiche (ereditarie) nell’agranulocitosi (Aprire la tabella in una nuova finestra)
|
Sindrome |
Inheritance |
Gene |
Clinical Features |
|
Cyclic neutropenia |
Autosomal dominant |
ELA2 |
Alternate 21-day cycling of neutrophils and monocytes |
|
Kostmann syndrome |
Autosomal recessive |
Unknown |
Stable neutropenia, no MDS or AML |
|
Severe congenital neutropenia |
Autosomal dominant |
ELA2 (35-84%) |
Stable neutropenia, MDS or AML |
|
Autosomal dominant |
GFI1 |
Stable neutropenia, circulating myeloid progenitors, lymphopenia |
|
|
Sex linked |
Wasp |
Neutropenic variant of Wiskott-Aldrich syndrome |
|
|
Autosomal dominant |
G-CSFR |
G-CSF–refractory neutropenia, no AML or MDS |
|
|
Hermansky-Pudlak syndrome type 2 |
Autosomal recessive |
AP3B1 |
Severe congenital neutropenia, platelet dense-body defect, oculocutaneous albinism |
|
Chediak-Higashi syndrome |
Autosomal recessive |
LYST |
Neutropenia, oculocutaneous albinism, giant lysosomes, impaired platelet function |
|
Barth syndrome |
Sex linked |
TAZ |
Neutropenia, often cyclic; cardiomyopathy, methylglutaconic aciduria |
|
Cohen syndrome |
Autosomal recessive |
COH1 |
Neutropenia, mental retardation, dysmorphism |
|
Source: Modified from Berliner et al, 2004. AML = acute myeloid leukemia; G-CSF = granulocyte colony-stimulating factor; MDS = myelodysplastic syndrome. |
|||
Le cause della neutropenia acquisita sono complesse, ma la maggior parte sono legate a tre categorie principali: infezioni, farmaci (sia tossici diretti che immunomediati) e autoimmunità. La neutropenia cronica benigna, o neutropenia cronica idiopatica, sembra essere un disturbo di sovrapposizione con le forme ereditarie e acquisite, e a volte è indistinguibile. Alcuni pazienti neutropenici danno un’anamnesi chiara e un modello familiare, mentre altri non hanno un’anamnesi familiare, poche determinazioni degli esami del sangue e una durata sconosciuta della neutropenia. Questo gruppo di pazienti potrebbe avere neutropenia ereditaria o acquisita. Un breve riassunto dei disturbi neutropenici sia congeniti che acquisiti segue.
Neutropenia con difetti immunitari associati
Neutropenia con immunoglobuline anormali si osserva in individui con agammaglobulinemia legata all’X, deficit isolato di immunoglobulina A (IgA), sindrome X-linked hyperimmunoglobulin M (XHIGM) e disgammaglobulinemia tipo I. Nella XHIGM, che è dovuta a mutazioni nel ligando CD40, i pazienti possono effettivamente avere livelli normali o elevati di IgM ma livelli sierici di IgG notevolmente diminuiti. In tutti questi disturbi, il rischio di infezione è alto, e il trattamento è l’immunoglobulina per via endovenosa (IVIG).
I pazienti con disgenesia reticolare dimostrano una grave neutropenia, nessuna immunità cellulo-mediata, agammaglobulinemia e linfopenia. Si verificano infezioni pericolose per la vita che sono refrattarie al fattore stimolante le colonie di granulociti (G-CSF). Il trapianto di midollo osseo è il trattamento di scelta.
Nutropenie congeniali o croniche
La neutropenia congenita grave (SCN), o sindrome di Kostmann, è più spesso causata da un’eredità recessiva e si trova in popolazioni remote e isolate con un alto grado di consanguineità. Sono stati riportati anche casi autosomici dominanti e sporadici, il più delle volte dovuti a mutazioni nel recettore del G-CSF. Non esiste un difetto genetico uniforme in questa sindrome. Le mutazioni in ELA2, che sono causative per la neutropenia ciclica (vedi sotto) non sono sufficienti a spiegare il fenotipo di Kostmann-like SCN.
I pazienti si presentano all’età di 3 mesi con infezioni batteriche ricorrenti. La bocca e il periretto sono i siti più comuni di infezione. Questo tipo di neutropenia è grave e il trattamento è G-CSF. Il rischio di conversione in sindrome mielodisplastica (SMD)/leucemia mieloide acuta (AML) con monosomia 7 dopo i trattamenti con G-CSF è associato a ulteriori mutazioni acquisite. La maggior parte di questi casi sono causati da una mutazione nel recettore del G-CSF. I pazienti la cui condizione risponde clinicamente al G-CSF sono trattati per tutta la vita.
Alcuni pazienti con altre forme di SCN sembrano avere mutazioni in GFI1, un gene repressore trascrizionale a zinco-finger coinvolto nella funzione delle cellule staminali ematopoietiche e nelle decisioni di impegno di linea.
La neutropenia ciclica (CN) è caratterizzata da attacchi periodici di neutropenia associati a infezioni, seguiti da un recupero della conta periferica dei neutrofili. La sua periodicità è di circa 21 giorni (range, 12-35 d). I precursori dei granulociti scompaiono dal midollo prima di ogni nadir di neutrofili nel ciclo a causa dell’apoptosi accelerata delle cellule progenitrici mieloidi. Alcuni casi possono essere determinati geneticamente con un’eredità autosomica recessiva. Altri casi possono essere dovuti a un’eredità autosomica dominante. In alcuni casi sporadici di CN, i pazienti hanno mutazioni in ELA2.
Le persone con CN si presentano tipicamente come neonati o bambini, ma esistono forme acquisite di CN in età adulta. La prognosi è buona, con un decorso benigno; tuttavia, il 10% dei pazienti sperimenterà infezioni pericolose per la vita. Il trattamento per la neutropenia ciclica è il G-CSF quotidiano.
Nutropenia cronica benigna
La neutropenia cronica benigna familiare, o neutropenia etnica benigna, è un disordine con un modello autosomico dominante di eredità osservato in africani, ebrei yemeniti, ebrei etiopi, arabi, caraibici e di origine indiana occidentale. Nelle popolazioni di discendenza africana ed ebrea yemenita, gli studi genetici mostrano una forte associazione con un polimorfismo a singolo nucleotide nel gene DARC. I pazienti sono tipicamente asintomatici e le infezioni sono lievi. Gli individui affetti da neutropenia benigna cronica hanno un rischio complessivamente basso di infezione e non è richiesta alcuna terapia specifica.
Nelle neutropenie croniche benigne non familiari, le infezioni lievi con un decorso benigno caratterizzano questo disturbo. L’ANC, tuttavia, risponde allo stress, come l’infezione, i corticosteroidi e le catecolamine.
Neutropenia cronica grave idiopatica
La neutropenia cronica grave idiopatica è una diagnosi di esclusione. I pazienti affetti presentano infezioni e neutropenia grave.
Neutropenia associata ad anomalie fenotipiche
La sindrome di Shwachman (Shwachman-Diamond) ha un modello ereditario autosomico recessivo. La neutropenia è da moderata a grave, con un tasso di mortalità del 15-25%, e la sindrome si presenta nell’infanzia, con infezioni ricorrenti, diarrea e difficoltà di alimentazione. Possono verificarsi nanismo, condrodisplasia e insufficienza pancreatica esocrina.
La sindrome di Shwachman-Diamond e la discheratosi congenita X-linked (DC), l’ipoplasia cartilage-hair (CHH) e l’anemia Diamond-Blackfan (DBA) sembrano condividere difetti genetici comuni coinvolti nella sintesi dei ribosomi. La maggior parte dei casi di sindrome di Shwachman-Diamond sono causati da mutazioni nel gene SBDS. La funzione precisa di questo gene è ancora in fase di chiarimento; tuttavia, è coinvolto nella sintesi dei ribosomi e nelle reazioni di elaborazione dell’RNA. Il trattamento è il G-CSF.
Nella CHH, il modello di eredità è autosomico recessivo sul cromosoma 9, e si osserva nelle famiglie amish e finlandesi. La CHH è causata da mutazioni nel gene RMRP, che codifica il componente RNA del complesso di elaborazione dell’RNA mitocondriale ribonucleasi (RNase MRP). La neutropenia è da moderata a grave. Il CHH si presenta con difetti dell’immunità cellulo-mediata, anemia macrocitica, malattia gastrointestinale e nanismo. Mostra anche una predisposizione al cancro, specialmente al linfoma. Il trattamento è il trapianto di midollo osseo.
La discheratosi congenita (sindrome di Zinsser-Cole-Engman) si presenta con ritardo mentale, pancitopenia e immunità cellulo-mediata difettosa. La discheratosi congenita è più comune negli uomini che nelle donne ed è ematologicamente simile all’anemia di Fanconi. La discheratosi congenita è solitamente recessiva X-linked, anche se esistono forme autosomiche dominanti e autosomiche recessive di questo disordine.
La forma recessiva X-linked del disordine è stata collegata a mutazioni in DKC1, che codifica la discherina, una proteina nucleolare associata a particelle ribonucleoproteiche. La forma autosomica dominante è associata a mutazioni in un altro gene, TERC, che fa parte della telomerasi. La telomerasi ha sia una componente proteica che una RNA, e TERC codifica la componente RNA. I pazienti con questo disturbo hanno telomeri più corti del normale. Il trattamento è G-CSF, fattore stimolante le colonie di granulociti-macrofagi (GM-CSF) e trapianto di midollo osseo.
La sindrome di Barth è un disordine X-linked recessivo che si presenta con cardiomiopatia nell’infanzia, miopatia scheletrica, infezioni ricorrenti, nanismo e neutropenia moderata o grave.
La sindrome di Chediak-Higashi è un disordine autosomico recessivo con infezioni ricorrenti, rallentamento mentale, fotofobia, nistagmo, albinismo oculocutaneo, neuropatia, disturbi di sanguinamento, gengivite e granuli lisosomiali in varie cellule. La neutropenia è da moderata a grave, e il trattamento è il trapianto di midollo osseo.
Myelokathexis
Myelokathexis si presenta nell’infanzia con neutropenia moderata ed è associato a infezioni ricorrenti. La condizione è dovuta all’apoptosi accelerata e alla diminuzione dell’espressione di bcl-x nei precursori dei neutrofili. Si osserva un aspetto nucleare anormale, con ipersegmentazione con filamenti nucleari, picnosi e vacuolizzazione citoplasmatica. Il trattamento è G-CSF e GM-CSF.
Sindrome dei leucociti pigri
La sindrome dei leucociti pigri è una grave neutropenia con associata motilità anormale dei neutrofili. L’eziologia è sconosciuta e il trattamento è di supporto.
Disordini metabolici
Sono neutropenie croniche con ANC variabili. They include glycogen storage disease type 1b and various acidemias, such as isovaleric, propionic, and methylmalonic. In glycogen storage disease type 1b, the treatment is G-CSF and GM-CSF.
Acquired neutropenia caused by intrinsic bone marrow disease
Intrinsic bone marrow diseases that may cause neutropenia include the following:
-
Aplastic anemia
-
Hematologic malignancy (eg, leukemia, lymphoma, myelodysplasia, myeloma)
-
Ionizing radiation
-
Tumor infiltration
-
Granulomatous infection
-
Myelofibrosis
Immune-mediated neutropenia
A drug may act as a hapten and induce antibody formation. This mechanism operates in cases due to gold, aminopyrine, and antithyroid drugs. The antibodies destroy the granulocytes and may not require the continued presence of the drug for their action. Alternatively, the drug may form immune complexes that attach to the neutrophils. This mechanism operates with quinidine.
Drug immune-mediated neutropenia may be caused by the following:
-
Aminopyrine
-
Quinidine
-
Cephalosporins
-
Penicillins
-
Sulfonamides
-
Phenothiazines
-
Hydralazine
Other medications have been implicated
Autoimmune neutropenia is the neutrophil analogue of autoimmune hemolytic anemia and of idiopathic thrombocytopenic neutropenia. It should be considered in the absence of any of the common causes. Antineutrophil antibodies have been demonstrated in these patients. Autoimmune neutropenia may be associated with the following:
-
Rheumatoid arthritis (with or without Felty syndrome)
-
Sjögren syndrome
-
Chronic, autoimmune hepatitis
-
Systemic lupus erythematosus
-
Thymoma
-
Goodpasture disease
-
Granulomatosis with polyangiitis (Wegener granulomatosis)
-
Pure red blood cell (RBC) aplasia, in which there is complete disappearance of granulocyte tissue from the bone marrow; pure RBC dysplasia is a rare disorder due to the presence of antibody-mediated, granulocyte-macrophage colony forming unit (GM-CFU) inhibitory activity, and it is often associated with thymoma
-
Transfusion reactions, which can be caused by the surface antigens of neutrophilia; I destinatari di ripetute trasfusioni di granulociti potrebbero diventare alloimmunizzati
-
Proliferazione di grandi linfociti granulari o leucemia
Nella neutropenia neonatale isoimmune, la madre produce anticorpi IgG antineutrofili contro antigeni neutrofili fetali che vengono riconosciuti come non self. Questo si verifica nel 3% dei nati vivi. Il disturbo si manifesta con febbre neonatale, infezioni del tratto urinario, cellulite, polmonite e sepsi. La durata della neutropenia è tipicamente di 7 settimane.
La neutropenia cronica autoimmune si osserva negli adulti e non ha una predilezione di età. Fino al 36% dei pazienti mostrerà anticorpi antineutrofili nel siero, e il decorso clinico è solitamente meno grave. I pazienti possono avere questo disturbo in associazione con lupus eritematoso sistemico, artrite reumatoide, granulomatosi di Wegener ed epatite cronica.
Se la neutropenia cronica autoimmune è associata a queste malattie, i corticosteroidi sono indicati come trattamento. Nei neonati e nei bambini, questo disordine è associato a un minor rischio di infezione e a infezioni più lievi che coinvolgono l’orecchio medio, il tratto gastrointestinale e la pelle.
La linfocitosi T-gamma, o disordine linfoproliferativo, è una malattia clonale dei linfociti T CD3+ o delle cellule natural killer (NK) CD3- che infiltrano il midollo osseo e i tessuti. Conosciuta anche come leucemia dei grandi linfociti granulari (LGL-leucemia), la linfocitosi T-gamma può essere associata all’artrite reumatoide ed è associata ad anticorpi antineutrofili ad alto titolo. La neutropenia è persistente e grave. Il trattamento è spesso di supporto in natura, ma è anche diretto ad eliminare la popolazione clonale.
Nutropenia acquisita causata da infezione
Le infezioni sono la forma più comune di neutropenia acquisita. Le infezioni che possono causare neutropenia includono, ma non sono limitate a quanto segue:
-
Bacterial sepsis
-
Viral infections (eg, influenza, measles, Epstein Barr virus , cytomegalovirus , viral hepatitis, human immunodeficiency virus -1) (see first image below)
-
Toxoplasmosis
-
Brucellosis
-
Typhoid
-
Tuberculosis (see second and third images below)
-
Malaria
-
Dengue fever
-
Rickettsial infection
-
Babesiosis
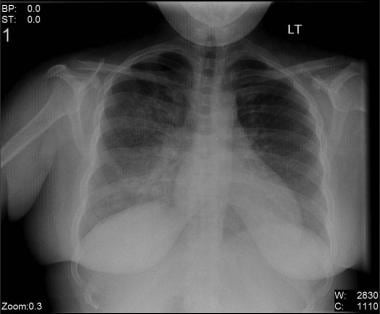 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia. 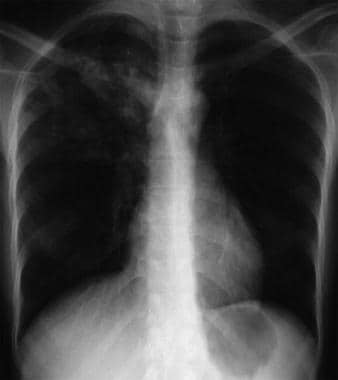 Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Questa donna è stata ricoverata in isolamento e iniziata empiricamente con un regime di 4 farmaci in ED. La tubercolosi è stata confermata dal test dell’espettorato. Immagine per gentile concessione di Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Questa donna è stata ricoverata in isolamento e iniziata empiricamente con un regime di 4 farmaci in ED. La tubercolosi è stata confermata dal test dell’espettorato. Immagine per gentile concessione di Remote Medicine, remotemedicine.org. 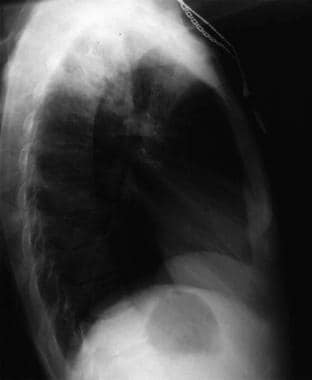 Radiografia laterale del torace in un paziente di 31 anni
Radiografia laterale del torace in un paziente di 31 anni Radiografia laterale del torace in un paziente di 31 anni con polmonite influenzale. Immagine per gentile concessione di Remote Medicine, remotemedicine.org.
 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.  Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Questa donna è stata ricoverata in isolamento e iniziata empiricamente con un regime di 4 farmaci in ED. La tubercolosi è stata confermata dal test dell’espettorato. Immagine per gentile concessione di Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Questa donna è stata ricoverata in isolamento e iniziata empiricamente con un regime di 4 farmaci in ED. La tubercolosi è stata confermata dal test dell’espettorato. Immagine per gentile concessione di Remote Medicine, remotemedicine.org.  Radiografia laterale del torace in un paziente di 31 anni
Radiografia laterale del torace in un paziente di 31 anni Gli organismi più comunemente coinvolti provengono dalla flora endogena. Gli organismi Staphylococcus aureus si trovano nei casi di infezioni della pelle. Gli organismi Gram-negativi si osservano nelle infezioni del tratto urinario e gastrointestinale, in particolare le specie Escherichia coli e Pseudomonas. Possono anche verificarsi infezioni da Candida albicans. Una flora mista può essere trovata nella cavità orale.
Le infezioni virali spesso portano a neutropenia lieve o moderata. L’agranulocitosi è rara ma può verificarsi. Gli organismi più comuni sono il virus di Epstein-Barr, il virus dell’epatite B, il virus della febbre gialla, il citomegalovirus e l’influenza. Molte infezioni travolgenti, sia virali che batteriche, possono causare grave neutropenia.
Nutropenia acquisita causata da carenze nutrizionali
Le carenze nutrizionali che possono causare neutropenia includono vitamina B-12, folati, e carenza di rame.
Nutropenia acquisita causata da farmaci e sostanze chimiche, esclusa la chemioterapia citotossica
Numerosi farmaci sono stati associati a neutropenia. Le categorie a più alto rischio sono i farmaci antitiroidei, i macrolidi e i procainamidi. Come detto sopra, molti farmaci agiscono attraverso un meccanismo immunomediato. Tuttavia, alcuni farmaci sembrano avere effetti tossici diretti sulle cellule staminali del midollo o sui precursori dei neutrofili nel compartimento mitotico. Per esempio, farmaci come gli antipsicotici e gli antidepressivi e il cloramfenicolo possono agire come tossine dirette in alcuni individui, in base al metabolismo e alla sensibilità in questo modo. Altri farmaci possono avere una combinazione di meccanismi immunitari e non immunitari o possono avere meccanismi d’azione sconosciuti.
Gli antimicrobici includono penicillina, cefalosporine, vancomicina, cloramfenicolo, gentamicina, clindamicina, doxiciclina, flucitosina, nitrofurantoina, novobiocina, minociclina, griseofulvina, lincomicina, metronidazolo, rifampicina, isoniazide, streptomicina, tiacetazone, mebendazolo, pirimetamina, levamisolo, ristocetina, sulfamidici, clorochina, idrossiclorochina, chinacrina, etambutolo, dapsone, ciprofloxacina, trimetoprim, imipenem/cilastatina, zidovudina, fludarabina, aciclovir e terbinafina.
Gli agenti analgesici e antinfiammatori includono aminopirina, dipirone, indometacina, ibuprofene, acido acetilsalicilico, diflunisal, sulindac, tolmetina, benoxaprofene, barbiturici, mesalazina e chinino.
Antipsicotici, antidepressivi e agenti neurofarmacologici includono fenotiazine (clorpromazina, metilpromazina, mepazina, promazina, tioridazina, prochlorperazine, trifluoperazine, trimeprazine), clozapine, risperidone, imipramine, desipramine, diazepam, clordiazepoxide, amoxapine, meprobamate, thiothixene, e haloperidol.
Gli anticonvulsivanti includono acido valproico, fenitoina, trimetadione, mefenitoina (Mesantoina), etosuimide e carbamazepina.
I farmaci antitiroidei includono tiouracile, propiltiouracile, metimazolo, carbimazolo, perclorato di potassio e tiocianato.
I farmaci cardiovascolari comprendono procainamide, captopril, aprindina, propranololo, idralazina, metildopa, chinidina, diazoxide, nifedipina, propafenone, ticlopidina e vesnarinone.
Gli antistaminici comprendono cimetidina, ranitidina, tripelennamina (piribenzamina), metafenilene, thenalidina, bromfeniramina e mianserina.
I diuretici comprendono acetazolamide, bumetanide, clorotiazide, idroclorotiazide, clortalidone, metazolamide e spironolattone.
Gli agenti ipoglicemici includono clorpropamide e tolbutamide.
I farmaci antimalarici includono amodiachina, dapsone, idrossiclorochina, pirimetamina e chinino.
Farmaci vari includono allopurinolo, colchicina, aminoglutetimide, famotidina, bezafibrato, flutamide, tamoxifene, penicillamina, acido retinoico, metoclopramide, fenindione, dinitrofenolo, acido etacrinico, diclorodifeniltricloroetano (DDT), cinofene, antimonio, piritildione, rauwolfia, etanolo, clorpropamide, tolbutamide, tiazidi, spironolattone, metazolamide, acetazolamide, IVIG e levodopa.
I metalli pesanti includono oro, arsenico e mercurio.
L’esposizione a farmaci o sostanze chimiche è la causa più comune di agranulocitosi: circa la metà dei pazienti ha una storia di esposizione a farmaci o sostanze chimiche. Qualsiasi prodotto chimico o farmaco che può deprimere il midollo osseo e causare ipoplasia o aplasia è in grado di causare agranulocitosi. Alcuni farmaci lo fanno a tutti se sono somministrati in dosi abbastanza grandi. Altri agenti sembrano causare reazioni idiosincratiche che colpiscono solo alcuni individui suscettibili.
Alcuni agenti (es. acido valproico, carbamazepina e antibiotici beta-lattamici) agiscono per inibizione diretta della mielopoiesi. Nelle colture di midollo osseo, questi agenti inibiscono la formazione di colonie di granulociti in modo correlato alla dose. Il danno diretto al microambiente del midollo osseo o ai precursori mieloidi gioca un ruolo nella maggior parte degli altri casi.
Molti farmaci associati all’agranulocitosi sono stati segnalati alla US Food and Drug Administration (FDA) sotto il suo requisito di segnalazione delle reazioni avverse. Molti agenti sono anche segnalati a un registro mantenuto dall’American Medical Association (AMA). I farmaci segnalati sono stati utilizzati da soli, in combinazione con un altro farmaco noto per essere potenzialmente tossico, o con un altro farmaco senza tossicità nota. Diversi farmaci sono particolarmente salienti a causa della loro alta frequenza di associazione con agranulocitosi. Essi includono i seguenti:
-
Phenothiazine
-
Antithyroid drugs (thiouracil and propylthiouracil)
-
Aminopyrine
-
Chloramphenicol
-
Sulfonamides
Miscellaneous immunologic neutropenias
Immunologic neutropenias may occur after bone marrow transplantation and blood product transfusions.
Felty syndrome is a syndrome of rheumatoid arthritis, splenomegaly, and neutropenia. Splenectomy shows an initial response, but neutropenia may recur in 10-20% of patients. Treatment is directed toward rheumatoid arthritis.
In complement activation–mediated neutropenia, hemodialysis, cardiopulmonary bypass, and extracorporeal membrane oxygenation (ECMO) expose blood to artificial membranes and can cause complement activation with subsequent neutropenia.
In splenic sequestration, the degree of neutropenia resulting from this process is proportional to the severity of the splenomegaly and the bone marrow’s ability to compensate for the reduction in circulating bands and neutrophils.
Eosinopenia and basophilopenia
Eosinopenia may be associated with the following:
-
Acute bacterial infection
-
Glucocorticoid administration
-
Physical stress
-
Thymoma
Decreased circulating basophils may be associated with the following:
-
Anaphylaxis
-
Acute infection
-
Drug-induced hypersensitivity
-
Congenital absence of basophils
-
Hemorrhage
-
Hyperthyroidism
-
Ionizing radiation
-
Neoplasia
-
Ovulation
-
Urticaria
-
Drugs (eg, corticosteroid, adrenocorticotropic hormone therapy, chemotherapeutic agents, thyroid hormones)
Go to Pediatric Autoimmune and Chronic Benign Neutropenia for complete information on this topic.