2.3D: Teori för separation
Allmän teori
TLC är ett utmärkt analytiskt verktyg för att separera blandningar i ett prov. I det här avsnittet diskuteras detaljerna i separationen, och utökar den allmänna diskussionen i avsnitt 2.1.B.
I alla former av kromatografi jämnar proverna ut sig mellan stationär och mobil fas. I nästan alla tillämpningar av TLC är den stationära fasen ett adsorbent av kiseldioxid eller aluminiumoxid och den rörliga fasen är ett organiskt lösningsmedel eller en lösningsmedelsblandning (”eluent”) som stiger uppåt på plattan (ekvation 3).
\\
Kiseldioxidgel (som visas i figur 2.16) är uppbyggd av ett nätverk av kisel-syrebindningar, med \(\ce{O-H}\)-bindningar på ytan, samt ett lager av vattenmolekyler. Silicagel \(\left( \ce{SiO_2} \cdot x \ce{H_2O} \right)\) används i denna diskussion, men är strukturellt analog med aluminiumoxid \(\left( \ce{Al_2O_3} \cdot x \ce{H_2O} \right)\). Denna mycket polära stationära fas paras ihop med en relativt opolär mobil fas (ett organiskt lösningsmedel eller en organisk lösning) i det som kallas ”normalfas” TLC. Även om detta är den vanligaste formen av TLC (och vad som kommer att fokuseras på i detta avsnitt), används ibland ”omvänd fas” TLC (med en opolär stationär fas och en polär mobil fas).
Figur 2.16 visar hur acetofenon skulle fästa vid kiselgelens yta genom intermolekylära krafter (IMF). I det här fallet kan acetofenon vätebindas (den IMF som anges i figur 2.16a) till kiseldioxidytan genom sin syreatom. När eluenten strömmar över provet (figur 2.16b) etableras en jämvikt mellan provet som adsorberas på den stationära fasen och löses upp i den rörliga fasen. I den rörliga fasen rör sig föreningen uppåt på plattan med vätskeflödet (figur 2.16c) för att senare adsorberas på den stationära fasen längre upp på plattan. Den resulterande \(R_f\) för föreningen är beroende av den tid som tillbringats i den stationära och mobila fasen.
.png?revision=1&size=bestfit&width=1110&height=395)
Jämviktsfördelningen mellan de två faserna beror på flera faktorer:
- Den beror på styrkan i de intermolekylära krafterna mellan provet och den stationära fasen.
En förening som bildar starka IMF:er med kiseldioxid eller aluminiumoxid kommer ofta att gynna den stationära fasen och kommer att tillbringa en stor del av elueringstiden vidhäftat till plattan. Detta innebär att den kommer att tillbringa mindre tid i den rörliga fasen (som är det enda sättet för den att ta sig uppför plattan), vilket gör att den hamnar lågt på TLC-plattan och har ett lågt \(R_f\).
Föreningar som har syre- eller kväveatomer bör kunna vätebindas med den stationära fasen (har starka IMF med den stationära fasen), och kommer därför att ha lägre \(R_f\)-värden än föreningar av liknande storlek som endast kan interagera genom Londons dispersionskrafter (LDF). - Det beror på styrkan i interaktionen mellan provet och den rörliga fasen.
Då den rörliga fasen alltid är mindre polär än den stationära fasen i normalfas-TLC kommer polära föreningar att ha en tendens att ha en mindre affinitet för den rörliga fasen än opolära föreningar (baserat på principen ”likadana ämnen löser likadana ämnen”). Därför tenderar polära föreningar att tillbringa mindre av elutionstiden i rörelse än en opolär förening och kommer därför att färdas ”långsammare” uppåt på plattan och ha en låg \(R_f\).
Den grad av attraktion som en förening har till den stationära och mobila fasen leder till samma slutsats:
- Desto starkare IMF är möjlig med den stationära fasen (ofta ju mer polära funktionella grupper på en förening), desto längre tid kommer föreningen att vara stationär \(\rightarrow\) lägre \(R_f\).
- Desto fler polära funktionella grupper som finns på en förening, desto mindre tenderar den att attraheras av den mindre polära eluenten, och desto kortare tid kommer föreningen att vara rörlig \(\rightarrow\) lägre \(R_f\).
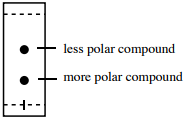
Tyvärr tenderar en förening med lägre \(R_f\) att ha fler polära funktionella grupper än en förening med högre \(R_f\) (sammanfattas i figur 2.17).