2.3D: Teorie dělení
Obecná teorie
TLC je vynikající analytický nástroj pro dělení směsí ve vzorku. V této části jsou probrány detaily separace a rozšířena obecná diskuse v části 2.1.B.
Ve všech formách chromatografie se vzorky vyrovnávají mezi stacionární a mobilní fází. Téměř ve všech aplikacích TLC je stacionární fází adsorbent z oxidu křemičitého nebo oxidu hlinitého a mobilní fází je organické rozpouštědlo nebo směs rozpouštědel („eluent“), která stoupá po desce (rovnice 3).
\
Silikagel (znázorněný na obrázku 2.16) je tvořen sítí vazeb mezi křemíkem a kyslíkem, přičemž na jeho povrchu se nacházejí vazby \(\ce{O-H}\) a také vrstva molekul vody. Silikagel \(\left( \ce{SiO_2} \cdot x \ce{H_2O} \right)\) se používá v této diskusi, ale strukturně je analogický oxidu hlinitému \(\left( \ce{Al_2O_3} \cdot x \ce{H_2O} \right)\). Tato velmi polární stacionární fáze je spojena s relativně nepolární mobilní fází (organické rozpouštědlo nebo roztok), což se označuje jako „normální fáze“ TLC. Ačkoli se jedná o nejběžnější formu TLC (na kterou se v této části zaměříme), někdy se používá i TLC s „reverzní fází“ (s nepolární stacionární fází a polární mobilní fází).
Obrázek 2.16 ukazuje, jak by acetofenon ulpíval na povrchu silikagelu prostřednictvím mezimolekulárních sil (IMF). V tomto případě se může acetofenon vázat vodíkovou vazbou (IMF naznačená na obrázku 2.16a) na povrch oxidu křemičitého prostřednictvím svého atomu kyslíku. Jak eluent proudí přes vzorek (obrázek 2.16b), nastává rovnováha mezi vzorkem adsorbovaným na stacionární fázi a rozpuštěným v mobilní fázi. Když je sloučenina v mobilní fázi, pohybuje se s proudem kapaliny vzhůru po desce (obrázek 2.16c), aby se později readsorbovala na stacionární fázi dále po desce. Výsledná hodnota \(R_f\) sloučeniny závisí na době strávené ve stacionární a mobilní fázi.
.png?revision=1&size=bestfit&width=1110&height=395)
Rovnovážné rozložení mezi oběma fázemi závisí na několika faktorech:
- Závisí na síle mezimolekulárních sil mezi vzorkem a stacionární fází.
Sloučenina, která tvoří silné IMF s oxidem křemičitým nebo hlinitým, bude často upřednostňovat stacionární fázi a stráví většinu elučního času přilepená na desce. To znamená, že stráví méně času v mobilní fázi (která je jediným prostředkem pro její pohyb po desce), což způsobí, že skončí nízko na TLC desce a bude mít nízkou hodnotu \(R_f\).
Sloučeniny, které mají atomy kyslíku nebo dusíku, by měly být schopny vodíkové vazby se stacionární fází (mají silné IMF se stacionární fází), a proto budou mít nižší hodnoty \(R_f\) než sloučeniny podobné velikosti, které mohou interagovat pouze prostřednictvím Londonových disperzních sil (LDF). - Záleží na síle interakce mezi vzorkem a mobilní fází.
Jelikož mobilní fáze je při TLC s normální fází vždy méně polární než stacionární fáze, polární sloučeniny budou mít tendenci mít menší afinitu k mobilní fázi než nepolární sloučeniny (na základě principu „podobné se rozpouští podobně“). Proto mají polární sloučeniny tendenci strávit menší část elučního času v mobilním prostředí než nepolární sloučeniny, takže se budou po desce pohybovat „pomaleji“ a budou mít nižší \(R_f\).
Stupeň přitažlivosti sloučeniny ke stacionární a mobilní fázi vede ke stejnému závěru:
- Čím silnější IMF je možné se stacionární fází (často čím více polárních funkčních skupin na sloučenině), tím více času bude sloučenina stacionární \(\rightarrow\) nižší \(R_f\).
- Čím více polárních funkčních skupin je na sloučenině přítomno, tím méně má tendenci být přitahována k méně polárnímu eluentu a tím kratší dobu bude sloučenina mobilní \(\rightarrow\) nižší \(R_f\).
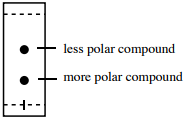
Sloučenina s nižším \(R_f\) má tedy tendenci mít více polárních funkčních skupin než sloučenina s vyšším \(R_f\) (shrnuto na obrázku 2).