2.3D: Teoria rozdzielania
Teoria ogólna
TLC jest doskonałym narzędziem analitycznym do rozdzielania mieszanin w próbce. W tej sekcji omówiono szczegóły rozdzielania i rozwinięto ogólną dyskusję z sekcji 2.1.B.
W każdej formie chromatografii, próbki wyrównują się pomiędzy fazą stacjonarną i ruchomą. W prawie wszystkich zastosowaniach TLC fazą stacjonarną jest adsorbent krzemionkowy lub glinowy, a fazą ruchomą rozpuszczalnik organiczny lub mieszanina rozpuszczalników („eluent”), która unosi się na płytce (równanie 3).
Żel krzemionkowy (pokazany na Rysunku 2.16) składa się z sieci wiązań krzemowo-tlenowych, z wiązaniami ∗(∗(∗(∗(∗(∗(∗(∗(∗(∗)∗) na jego powierzchni, a także z warstwy cząsteczek wody. W tej dyskusji używany jest żel krzemionkowy, ale jest on strukturalnie analogiczny do tlenku glinu. Ta bardzo polarna faza stacjonarna jest połączona ze względnie niepolarną fazą ruchomą (rozpuszczalnikiem organicznym lub roztworem), co jest określane jako „normalna faza” TLC. Chociaż jest to najbardziej powszechna forma TLC (i na niej skupimy się w tym rozdziale), czasami stosuje się TLC „fazy odwróconej” (z niepolarną fazą stacjonarną i polarną fazą ruchomą).
Rysunek 2.16 pokazuje, jak acetofenon przylgnąłby do powierzchni żelu krzemionkowego dzięki siłom międzycząsteczkowym (IMF). W tym przypadku acetofenon może wiązać się wodorem (IMF wskazany na Rysunku 2.16a) z powierzchnią krzemionki poprzez swój atom tlenu. Podczas przepływu eluentu nad próbką (Rysunek 2.16b) ustala się równowaga pomiędzy próbką zaadsorbowaną na fazie stacjonarnej i rozpuszczoną w fazie ruchomej. Po znalezieniu się w fazie ruchomej związek przemieszcza się w górę płytki wraz z przepływającą cieczą (rysunek 2.16c), aby później ulec adsorpcji na fazie stacjonarnej w dalszej części płytki. Wynikowa wartość R związku zależy od ilości czasu spędzonego w fazie stacjonarnej i ruchomej.
.png?revision=1&size=bestfit&width=1110&height=395)
Rozkład równowagi między dwiema fazami zależy od kilku czynników:
- Zależy od siły sił międzycząsteczkowych między próbką a fazą stacjonarną.
Związek, który tworzy silne IMF z krzemionką lub tlenkiem glinu, będzie często faworyzował fazę stacjonarną i spędzi dużą część czasu elucji przylegając do płytki. Oznacza to, że będzie spędzać mniej czasu w fazie ruchomej (która jest jedynym środkiem do przemieszczania się w górę płytki), powodując, że kończy się nisko na płytce TLC i ma niską wartość R. Związki, które mają atomy tlenu lub azotu powinny być w stanie wiązać wodór z fazą stacjonarną (mają silne IMF z fazą stacjonarną), a zatem będą miały niższe wartości R niż związki o podobnej wielkości, które mogą oddziaływać tylko poprzez londyńskie siły rozpraszające (LDF). - Zależy to od siły interakcji między próbką a fazą ruchomą.
Jako że faza ruchoma jest zawsze mniej polarna niż faza stacjonarna w normalnej fazie TLC, związki polarne będą miały tendencję do mniejszego powinowactwa do fazy ruchomej niż związki niepolarne (w oparciu o zasadę „podobne rozpuszcza się jak”). Dlatego związki polarne mają tendencję do spędzania mniejszej części czasu elucji w ruchu niż związki niepolarne, więc będą się przemieszczać „wolniej” w górę płytki i będą miały niski współczynnik \(R_f\).
Stopień przyciągania przez związek do fazy stacjonarnej i ruchomej prowadzi do tego samego wniosku:
- Im silniejsze IMF jest możliwe z fazą stacjonarną (często bardziej polarne grupy funkcjonalne na związku), tym więcej czasu związek będzie stacjonarny \(R_f\) niższe \(R_f\).
- Im więcej polarnych grup funkcyjnych obecnych na związku, tym mniejsza tendencja do bycia przyciągniętym do mniej polarnego eluentu i tym mniej czasu związek będzie ruchomy \(\rightarrow \) niższy \(R_f\).
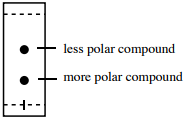
Tak więc, związek z niższym \(R_f\) ma tendencję do posiadania bardziej polarnych grup funkcyjnych niż związek z wyższym \(R_f\) (podsumowane na Rysunku 2.17).