Frontiers in Neurology
Wprowadzenie
Bustronne poziome porażenie wzroku jest rzadką postacią spowodowaną obustronnym przerwaniem powięzi podłużnej przyśrodkowej, jądra abducens lub paramedialnego tworu siatkowatego pontyny (PPRF) (1, 2). Rodzaje ruchów gałek ocznych, które mogą być zaburzone w uszkodzeniach pnia mózgu, obejmują poziome i pionowe wolne ruchy gałek ocznych, ruchy gonitwy, odpowiedzi przedsionkowe i optokinetyczne oraz szybkie ruchy gałek ocznych, w tym dobrowolne lub wywołane sakkady oraz szybkie fazy bodźców przedsionkowych i optokinetycznych. Na kontrolę wergencji mogą wpływać uszkodzenia śródmózgowia.
Paraliż spojrzenia horyzontalnego opisywano jako konsekwencję zawału (3), zapalenia/demielinizacji w ramach stwardnienia rozsianego (SM) lub spektrum zaburzeń neuromyelitis optica (NMOSD) (2, 4, 5), krwotoku (6) i przerzutów (7). Spośród opisanych wcześniej przypadków niezwiązanych z naczyniami/nowotworami, pięć z sześciu miało obustronne poziome porażenie wzroku, a trzy miały pojedyncze zmiany w obrazie rezonansu magnetycznego (MRI), zlokalizowane w części przyzwojowej. Milea i wsp. (1) opisali dwa przypadki obustronnej oftalmoplegii wewnątrzjądrowej (INO) przekształcającej się w obustronne poziome porażenie wzroku z pojedynczymi zmianami wzmacniającymi T2 w tej samej lokalizacji, o takiej samej morfologii jak nasza seria przypadków. Sugerowali oni MS jako prawdopodobną diagnozę.
Henderson opisał pacjenta z poziomą diplopią na lewym bocznym spojrzeniu i osłabieniem przywodzenia (8). MRI mózgu wykazało samotną hiperintensywną zmianę T2 pnia mózgu przylegającą do komory czwartej i postawiono diagnozę stwardnienia rozsianego. Wobec braku dodatkowych informacji klinicznych trudno jest ustalić, w jaki sposób postawiono to rozpoznanie. Matsui i wsp. opisali przypadek pacjenta skarżącego się na diplopię poziomą z obustronnym niedowładem spojrzenia poziomego (9). Postawiono diagnozę zapalenia pnia mózgu Bickerstaffa (BBE), chociaż cechy kliniczne nie były zgodne z kryteriami diagnostycznymi (10).
Opisujemy tu trzech pacjentów z obustronnym poziomym niedowładem wzroku, spowodowanym w każdym przypadku przez pojedynczą, zapalną zmianę obejmującą grzbietową część pnia mózgu, rozciągającą się ku przodowi do okolicy powięzi podłużnej średniej (MLF). Wierzymy, że te trzy przypadki podkreślają pytania dotyczące patogenezy takich zespołów i relacji poziomego i pionowego spojrzenia związanego z projekcją do MLF w szczególności.
Zgodnie z wymogami etyki naszej instytucji, Royal Melbourne Hospital, uzyskano świadomą, pisemną zgodę na wykorzystanie anonimowych danych od wszystkich pacjentów.
Pacjent 1
38-letni mężczyzna pochodzenia tureckiego przedstawił się z ostrą przerywaną diplopią, zawrotami głowy i wymiotami; zdiagnozowano u niego obwodowy zawrót głowy i został wypisany. Tydzień później zgłosił się ponownie z lewostronnym drętwieniem twarzy. Wywiad lekarski, badanie układu nerwowego i wywiad rodzinny nie wykazały nieprawidłowości.
Ostrość wzroku wynosiła 0,63 OU, mierzona w skali dziesiętnej. Średnica źrenicy wynosiła 5 mm obustronnie i nie była reaktywna. Badanie motoryki oka (Tabela 1) ujawniło całkowitą niewydolność wszystkich poziomych ruchów gałek ocznych, dobrowolnych sakkad, płynnego pościgu, ruchów przedsionkowych i optokinetycznych. Pionowe sakkady, gładki pościg i ruchy przedsionkowe były nienaruszone do badania klinicznego, ale to nie wyklucza możliwości niewielkiego spowolnienia, które wymagałoby oculografii do identyfikacji. Badanie neurologiczne było poza tym normalne.
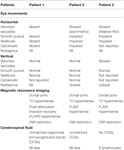
Tabela 1. Podsumowanie klinicznych ustaleń okulomotorycznych, wyników obrazowania MRI i wyników płynu mózgowo-rdzeniowego.
MRI mózgu wykazało hiperintensywną trójkątną zmianę T2 i fluid-attenuated inversion recovery (FLAIR) w obrębie grzbietowej części pons w dnie czwartej komory (Figura 1A). Zmiana wykazywała łagodnie ograniczoną dyfuzję dopasowaną do mapowania pozornego współczynnika dyfuzji, zgodną z doniesieniami w literaturze jako występującą ze zmianami zapalnymi/demielinizacyjnymi (11, 12), i nie wykazywała wzmocnienia kontrastowego. Rezonans magnetyczny mózgu i kręgosłupa był poza tym nieistotny. Stwierdzono, że zmiana w pniu mózgu ma cechy demielinizacji, chociaż nie zaobserwowano dalszych zmian. Choroba Behçeta, zapalenie naczyń lub zmiana mitotyczna były rozważane jako rozpoznania różnicowe.
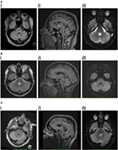
Rysunek 1. (A) I. Pacjent 1, osiowe badanie fluid-attenuated inversion recovery (FLAIR) ukazujące hiperintensywną zmianę w tylnej części opuszki przypontynowej. II. Saggitalny FLAIR ukazujący pionowy zasięg zmiany. III. Obrazowanie dyfuzyjne wykazujące łagodne ograniczenie dyfuzji. (B) I. Pacjent 2, osiowy podwójny obraz hiperintensywnej zmiany w tylnym przyzębiu. II. Saggitalny FLAIR ukazujący pionowy zasięg zmiany. III. Obrazowanie dyfuzyjne wykazujące łagodne ograniczenie dyfuzji. (C) I. Pacjent 3, flair osiowy uwidaczniający hiperintensywną zmianę w tylnym przyzębiu, nieco bardziej znaczącą po stronie prawej. II. Saggitalny FLAIR ukazujący pionowy zasięg zmiany, nieco większy niż u dwóch poprzednich pacjentów, zgodny z pewnym spowolnieniem pionowych sakkad. III. Obrazowanie dyfuzyjne pokazujące łagodne ograniczenie dyfuzji.
Analiza płynu mózgowo-rdzeniowego (CSF) ujawniła trzy limfocyty, brak polimorficznych i czerwonych krwinek, oligoklonalne pasma immunoglobulin (OCBs) były widoczne w CSF i nie były dopasowane w surowicy. Stwierdzono minimalnie podwyższony poziom białka (0,46 g/l), ale badania na obecność anty-akwaporyny 4 (Aq4) były negatywne. Surowica Aq4 była również ujemna. Testy na przeciwciała przeciwko gangliozydom nie zostały wykonane, a przeciwciała przeciwko glikoproteinie oligodendrocytów mieliny (MOG) nie były dostępne w czasie ostrej choroby, ale były negatywne, kiedy stały się dostępne 2 lata później.
Pacjent był leczony z powodu przypuszczalnej demielinizacji i jego stan poprawił się po 3 dniach podawania 1 g dożylnego metyloprednizolonu. Jego badanie neurologiczne powróciło do normy po 3 tygodniach od wypisu. Seryjne MRI mózgu wykazały całkowite ustąpienie zmian w opuszce, bez nowych zmian i bez dalszych epizodów klinicznych w ciągu 2 lat od pierwotnej prezentacji.
Pacjent 2
46-letnia kobieta z czołowym bólem głowy była leczona z powodu migreny i początkowo zareagowała na naproksen i amitryptylinę. Sześć dni później zgłosiła się ponownie z nasilającym się bólem głowy, światłowstrętem, nieostrym widzeniem, przerywaną poziomą diplopią i zmiennym drętwieniem twarzy.
Historia choroby i wywiad rodzinny były bez zmian. Badanie neurologiczne wykazało ostrość wzroku 0,32 OU. Reakcje źrenic były nieznacznie spowolnione, ale reaktywne obustronnie. Badanie okulomotoryczne przedstawiono w tabeli 1. Stwierdzono asymetryczny obustronny niedowład spojrzenia poziomego, prawostronny gorszy niż lewostronny. Sakkady w prawo miały ograniczoną amplitudę i były powolne. Odpowiedzi przedsionkowe były asymetryczne, a ruchy gałek ocznych wywołane manewrem głowy lalki były prawidłowe w lewym oku, ale ograniczone w prawym. Bardziej aktywne odpowiedzi pchnięcia głowy były hipoaktywne obustronnie. Oczopląs w górę był najbardziej widoczny przy patrzeniu w górę, ale przy oftalmoskopii zauważono go również w pozycji podstawowej. Pionowe sakkady, w górę i w dół, były normalne. Pionowy płynny pościg wydawał się nienaruszony. Pionowe uderzenia głową i odpowiedzi optokinetyczne były klinicznie normalne. Wideo okulografia nie była dostępna i, ogólnie, wyniki te zostały zinterpretowane jako zgodne z łagodnym upośledzeniem pionowego spojrzenia. Oczopląs pionowy ustąpił w ciągu 2 dni. Pacjentka miała również łagodne obustronne osłabienie twarzy, nieobecne odruchy ścięgniste w kończynach górnych i łagodną ataksję chodu. Wrażenie kliniczne było takie, że jej konstelacja objawów była zgodna z zespołem Millera Fishera, BBE lub centralną demielinizacją, ale bez dowodów potwierdzających rozpoznanie tych jednostek w testach.
Reonans magnetyczny mózgu ujawnił samotne, dobrze zdefiniowane paramedialne ognisko hiperintensywności T2 w grzbietowej części móżdżku, które wykazywało łagodnie ograniczoną dyfuzję, ale bez wzmocnienia kontrastowego (rysunek 1B). Badanie MRI kręgosłupa było prawidłowe.
Badanie przewodnictwa nerwowego ujawniło obustronną aksonalną neuropatię ruchową twarzy. Analiza płynu mózgowo-rdzeniowego wykazała OCBs (niedopasowane) i podwyższone stężenie albuminy (298 mg/l). Przeciwciało Aq4 w surowicy było ujemne. Rutynowe wyniki badania płynu mózgowo-rdzeniowego były prawidłowe, w tym białko 0,4 g/l, liczba białych krwinek (WCC) i liczba czerwonych krwinek (RCC) < 1. Płyn mózgowo-rdzeniowy był negatywny dla przeciwciał anty-AQP4, anty-GM1, anty-GQ1B i anty-białko mieliny-oligodendrocytów.
Diagnozą było zlokalizowane zapalenie mózgu ograniczone do pons i głównie wpływające na grzbietowe tegmentum. Pacjent był leczony 5-dniowym kursem dożylnej immunoglobuliny w dawce 2 g/kg. W ciągu następnego tygodnia oftalmoplegia uległa znacznej poprawie, poprawiły się odruchy z kończyn górnych i ustąpiła ataksja. W ciągu 12 miesięcy obserwacji nie stwierdzono żadnych nowych objawów ani zmian. Przegląd po 18 miesiącach potwierdził całkowite wyzdrowienie kliniczne.
Pacjent 3
Ten 29-letni mężczyzna obudził się z potami i dreszczami, a następnie z dwubocznym bólem głowy, zawrotami głowy, nudnościami i wymiotami. Podczas prezentacji 12 godzin później miał poziomą diplopię i prawostronne drętwienie. Ujawnił 5-dniowy wywiad dotyczący objawów ze strony górnych dróg oddechowych, zatkane zatoki oraz przebyte szczepienie przeciwko krztuścowi 1 miesiąc przed wystąpieniem objawów. W wywiadzie stwierdzono nadciśnienie tętnicze, niedoczynność tarczycy, lęk, zespół jelita drażliwego, otyłość i niealkoholowe stłuszczeniowe zapalenie wątroby.
Wykonany rezonans magnetyczny mózgu ujawnił lewostronną, niewzmacniającą się, hiperintensywną zmianę T2 w grzbietowej części mostu, która wykazywała ograniczoną dyfuzję (ryc. 1C). W płynie mózgowo-rdzeniowym stwierdzono podwyższony poziom WCC wynoszący 8 × 106/l (7 limfocytów, 1 neutrofil), RCC wynosił 4 × 106/l, a białko w płynie mózgowo-rdzeniowym wynosiło 0,35 g/l. Początkowo w innym ośrodku rozpoznano udar niedokrwienny mózgu i rozpoczęto leczenie w ramach prewencji wtórnej. Nie stwierdzono nieprawidłowości w badaniu angiograficznym rezonansu magnetycznego, monitorowaniu serca, echokardiogramie przezprzełykowym, badaniach w kierunku zapalenia naczyń i trombofilii.
Dwa tygodnie po wystąpieniu choroby u chorego wystąpiły zmiany smaku po lewej stronie, drętwienie lewego języka, wewnętrznej strony jamy ustnej i twarzy. Jego jednooczna ostrość wzroku wynosiła 1,0 OU, ale dynamiczna ostrość wzroku wynosiła 0,4 OU. Reakcje źrenic były prawidłowe. Wykazał obustronny niedowład spojrzenia i INO (Tabela 1). Reakcje na ruchy głowy były obustronnie hipoaktywne, z łagodnie spowolnionymi sakkadami w górę i oczopląsem przy podnoszeniu wzroku. Reszta badania neurologicznego ujawniła tylko łagodną lewostronną dysmetrię.
Powtórna analiza płynu mózgowo-rdzeniowego ujawniła 3 leukocyty (niezróżnicowane), 27 krwinek czerwonych i negatywne dla OCBs. Niekontrastowy rezonans magnetyczny mózgu ujawnił powiększenie zmiany z utrzymującym się ograniczeniem dyfuzji. Objawy były zgodne z demielinizacją. Przeciwciała antygangliozydowe, przeciwciała antyaquaporin-4 (AQP4) i przeciwciała anty-MOG w surowicy były ujemne.
Pacjent był leczony 3-dniowym kursem 1 g dziennie metyloprednizolonu dożylnie, a następnie 60 mg prednizolonu doustnie dziennie, powoli odstawianego po 1 miesiącu. Badanie kontrolne po 8 tygodniach od pierwszej prezentacji wykazało resztkowy lewy INO i tylko prawe objawy czuciowe, które ustąpiły w ciągu kolejnych miesięcy. Zmiana ustąpiła całkowicie w kontrolnych badaniach obrazowych 3 miesiące później.
Dyskusja
Opisujemy trzy przypadki obustronnego poziomego niedowładu wzroku, zbiegającego się z ostrym rozwojem małej, dobrze odgraniczonej zmiany w przyzębiu w badaniu MRI. Z definicji, przypadki te reprezentują klinicznie izolowane zespoły, ale przy braku nawracających zmian w 12-36 miesięcznej obserwacji. Fenotyp ten jest opisywany w MS, NMOSD, udarze mózgu i nowotworach (1, 4, 5, 8, 9, 13, 14). Jednak u chorych z tymi rozpoznaniami obserwuje się zwykle nawroty i charakterystyczne zmiany w MRI (4, 15). W naszych przypadkach nie ma wystarczających dowodów na potwierdzenie któregokolwiek z tych rozpoznań w momencie prezentacji lub w trakcie obserwacji. Skupiamy się tutaj na proponowanych nowościach klinicznych i anatomicznych, które demonstrują.
Zaburzenia ze spektrum anty-GQ1b mogą wpływać na ruchy gałek ocznych i potencjalnie mogą występować z opisanymi przez nas cechami klinicznymi. Zespół nakładania Miller-Fisher/BBE był rozważany w drugim przypadku, ale ci pacjenci mają tendencję do prezentowania z pozytywnymi przeciwciałami anty-GQ1B i normalnym MRI mózgu (13). W związku z tym uważamy, że bardziej prawdopodobna jest diagnoza alternatywna. Chociaż NMOSD może prezentować się ze zmianami zapalnymi w pobliżu przypontynowego tegmentum, są one charakterystycznie nieco niższe na dnie czwartej komory. Opisywane tu przypadki nie spełniają ostatnio zaktualizowanych kryteriów dla NMOSD (16).
Podobnie zespół CLIPPERS jest zaburzeniem zapalnym pnia mózgu, ale klinicznie i radiologicznie jest bardziej rozproszony i mało prawdopodobne, aby był wyjaśnieniem u naszych pacjentów (17).
Kontrola pionowego spojrzenia jest ogólnie przypisywana rostralnej części śródmózgowia, rostralnemu jądru śródmiąższowemu MLF (riMLF) zawierającemu komórki pękające oraz śródmiąższowemu jądru Cajala tworzącemu neuronalny integrator dla utrzymania pozycji w oczodole. Obszary te rzutują się obustronnie, częściowo przez zachyłek tylny. Wejścia do tych obszarów pochodzą z wielu źródeł, częściowo zależnych od typu wejścia.
Wejścia przedsionkowe pochodzą z jąder przedsionkowych górnych obustronnie, podczas gdy sygnały gładkiego pościgu rzutują z grupy komórek Y do riMLF. Ścieżki obejmują MLF, przewód brzuszno-zwieraczowy i drogi móżdżkowe. Dobrowolne i odruchowe sakkady pionowe, determinowane przez sieci nadnamiotowe, wydają się być zależne od projekcji z regionu PPRF obustronnie i są zniesione przez zlokalizowane uszkodzenia tylko ogonowego PPRF obustronnie (18). Wykazano silną projekcję neuronów omnipauzalnych z PPRF, obustronnie, do riMLF (19, 20). Muszą one być zahamowane, aby mogły być generowane dobrowolne i refleksyjne sakkady, a nie szybkie fazy (21).
Pionowe spojrzenie może być zachowane w obecności poziomej oftalmoplegii z uszkodzeniem tylnej części przypontyny z lub bez bardziej rozległej patologii (1, 13). Chociaż zmiany czysto opuszkowe mogą powodować poziome porażenie wzroku, w przypadkach opisanych przez Milea i wsp. wcześnie wystąpiło obustronne INO, co sugeruje zajęcie MLF lub obustronnych projekcji do MLF.
Ujednolicającą zmianą, u naszych pacjentów i tych opisanych tutaj, jest zmiana w tylnej części opuszki. Zmiana ta znajduje się w bliskim sąsiedztwie jąder abducens i PPRF zawierającego pobudzające komórki rozruchowe dla spojrzenia poziomego. Obustronne zajęcie projekcji z PPRF lub jąder abducens tłumaczyłoby porażenie spojrzenia poziomego. Względne oszczędzanie pionowych ruchów oczu sugeruje, że obustronne sygnały pionowe rzutowane do ośrodka pionowego spojrzenia formacji siateczkowo-mezencefalicznej przez MLF (22), biegną odrębną ścieżką w pons, w porównaniu z wejściami spojrzenia bocznego. Jest to zgodne z dowodami Pierrot-Deseilligny i wsp. (7), dotyczącymi obustronnego porażenia spojrzenia poziomego, ale sprzeczne z obecnym konwencjonalnym klinicznym postrzeganiem anatomii w tym obszarze.
Jeżeli występują obustronne uszkodzenia bocznych ośrodków spojrzenia w PPRF, blisko związanych i prawie nieodróżnialnych od jądra abdukcyjnego, konwencjonalna mądrość wskazywałaby, że powinny występować również porażenia spojrzenia pionowego, jako że mówi się, że te drogi biegną razem. Z pewnością zmiany w linii pośrodkowej dotyczące obu powięzi podłużnych środkowych (MLF) powodują obustronne INO, które jest charakterystycznie związane z pionowym niedowładem wzroku, charakteryzującym się upośledzeniem płynnego pościgu w pionie i zniesieniem odruchu przedsionkowo-ocznego (23). Zmiany bardziej rostralne mogą dotyczyć struktur śródmózgowia (riMLF i INC) zaangażowanych w sakkady pionowe (24). Sugerujemy, że pionowe i poziome drogi wzrokowe biegną oddzielnie przez ich dolny bieg i łączą się w ich rostralnym przebiegu, z pionowymi wejściami wzrokowymi biegnącymi przednio-bocznie do wejść wzrokowych poziomych. Mogą one przekroczyć w ich wznoszenia, prawdopodobnie znacznie, tak, że dwustronne zmiany są wymagane do wytworzenia pionowego porażenia spojrzenia. Wyjaśniałoby to oszczędzanie bezpośredniej drogi dla pionowego spojrzenia z bocznego PPRF w obecności jednostronnej zmiany, zniesienie pionowych ruchów gałek ocznych przy obustronnie zlokalizowanych zmianach oraz wymóg obustronnej stymulacji, aby sprowokować pionowe ruchy gałek ocznych (18, 22).
Proponujemy nieco zmieniony układ anatomiczny, aby to przedstawić (rysunek 2). Ścieżki spojrzenia poziomego rzutują się do ipsilateralnego jądra abducensa, a następnie wznoszą się w MLF do kontralateralnego podjądrza prostego przyśrodkowego jądra trzeciego nerwu. Proponujemy, że pionowy sygnał spojrzenia rzutuje się bardziej do przodu w MLF i, przynajmniej na krótkim dystansie, jest możliwy do oddzielenia od poziomych ścieżek włókien spojrzenia.
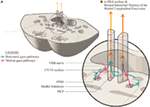
Figura 2. (A) Przekrój przez śródmózgowie pokazujący związek paramedialnego tworu siatkowatego pontyny z powięzią podłużną środkową (MLF) i przyległym siódmym nerwem czaszkowym, jak omija jądro abducens. (B) Powiększenie pokazujące proponowaną konformację oddzielnych poziomych i pionowych ścieżek spojrzenia, gdy rzutują się one na MLF. Sygnał horyzontalny rozpada się na poziomie pons i wznosi się w kontralateralnym MLF, podczas gdy nie jest pewne, czy pionowy sygnał spojrzenia rozpada się w ogóle, czy jest bezpośrednią projekcją.
Podobieństwa w fenotypie i morfologii zmian przyczynowych w tych przypadkach sugerują, że ten region przypontynowego tegmentum może mieć szczególne cechy czyniące go podatnym na atak immunologiczny. Żaden z tych przypadków nie spełnia kryteriów innych chorób, zapalnych lub innych, związanych z zajęciem MLF.
Jeśli rzeczywiście te przypadki reprezentują nowy fenotyp, to istnieje potrzeba rozeznania optymalnego postępowania, zarówno w ostrej fazie, jak i w dłuższym okresie. Błędna diagnoza, na przykład w przypadku stwardnienia rozsianego lub udaru mózgu, wiąże się z ryzykiem narażenia pacjenta na niepotrzebne długotrwałe leczenie przeciwpłytkowe lub immunomodulacyjne oraz na niepokój związany z rozpoznaniem istotnego, potencjalnie nawracającego, długotrwałego schorzenia z towarzyszącym niekorzystnym wpływem na jego wybory i możliwości życiowe.
Identyfikacja innych przypadków tego fenotypu byłaby przydatna, aby ułatwić poszukiwanie specyficznych nowych przeciwciał, które mogą tu wchodzić w grę, oraz aby wyjaśnić najbardziej odpowiedni i skuteczny przebieg leczenia.
Wkład Autora
RE dokonał przeglądu dokumentacji pacjenta i badań obrazowych, zebrał dane i skonstruował wstępne prezentacje, jak również przyczynił się do bieżącej edycji ostatecznej wersji przedłożonego manuskryptu. OW nadzorował RE w zbieraniu danych i był zaangażowany we wszystkie poziomy pisania i interpretacji danych, jak również w przegląd i edycję ostatecznej wersji manuskryptu. AB współpracowała z RE przy analizie danych, przeglądzie odpowiedniej literatury, interpretacji wyników i przygotowaniu manuskryptu.
Oświadczenie o konflikcie interesów
Niniejsze badania nie były wspierane finansowo przez żadne interesy komercyjne. Nie istnieją żadne relacje finansowe, które mogłyby być interpretowane jako potencjalny konflikt interesów.
1. Milea D, Napolitano M, Dechy H, Le Hoang P, Delattre J-Y, Pierrot-Deseilligny C. Kompletne obustronne poziome porażenie spojrzenia ujawniające stwardnienie rozsiane. J Neurol Neurosurg Psychiatry (2001) 70(2):252-5. doi:10.1136/jnnp.70.2.252
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Tan E, Kansu T. Bilateral horizontal gaze palsy in multiple sclerosis. J Clin Neuroophthalmol (1990) 10(2):124–6.
Google Scholar
3. Kataoka S, Hori A, Shirakawa T, Hirose G. Paramedian pontine infarction. Stroke (1997) 28(4):809–15. doi:10.1161/01.STR.28.4.809
CrossRef Full Text | Google Scholar
4. Furutani Y, Hata M, Miyamoto K, Moribata Y, Yoshimura N. A case of neuromyelitis optica masquerading as Miller Fisher syndrome. Case Rep Neurol (2014) 6(3):226–31. doi:10.1159/000368183
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Ng AW, Teoh SC. Dengue eye disease. Surv Ophthalmol (2015) 60(2):106–14. doi:10.1016/j.survophthal.2014.07.003
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Koehler PJ, Wattendorff AR, Goor C, Hekster REM, Falke THM. Bilateral horizontal gaze paralysis due to pontine hemorrhage. Clin Neurol Neurosurg (1986) 88(2):121–5. doi:10.1016/S0303-8467(86)80007-5
CrossRef Full Text | Google Scholar
7. Pierrot-Deseilligny C, Goasguen J, Chain F, Lapresle J. Pontine metastasis with dissociated bilateral horizontal gaze paralysis. J Neurol Neurosurg Psychiatry (1984) 47(2):159–64. doi:10.1136/jnnp.47.2.159
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Henderson S. A young patient with visual disturbance. Eur J Intern Med (2014) 25(6):e69–70. doi:10.1016/j.ejim.2014.05.007
CrossRef Full Text | Google Scholar
9. Matsui TA, Mineta H, Shioyama S, Kihara M, Takahashi M. . Clin Neurol (1998) 38(2):150–3.
Google Scholar
10. Bickerstaff ER. Brain-stem encephalitis; further observations on a grave syndrome with benign prognosis. Br Med J (1957) 1(5032):1384–7. doi:10.1136/bmj.1.5032.1384
CrossRef Full Text | Google Scholar
11. Schaefer PW, Grant PE, Gonzalez RG. Diffusion-weighted MR imaging of the brain. Radiology (2000) 217(2):331–45. doi:10.1148/radiology.217.2.r00nv24331
CrossRef Full Text | Google Scholar
12. Küker W, Ruff J, Gaertner S, Mehnert F, Mader I, Nägele T. Modern MRI tools for the characterization of acute demyelinating lesions: value of chemical shift and diffusion-weighted imaging. Neuroradiology (2004) 46(6):421-6. doi:10.1007/s00234-004-1203-5
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Kipfer S, Crook DW. Isolated bilateral horizontal gaze palsy as first manifestation of multiple sclerosis. Mult Scler J (2014) 20(6):754-5. doi:10.1177/1352458513518627
CrossRef Full Text | Google Scholar
14. Bronstein AM, Rudge P, Gresty MA, Du Boulay G, Morris J. Abnormalities of horizontal gaze. Clinical, oculographic and magnetic resonance imaging findings. II. Gaze palsy and internuclear ophthalmoplegia. J Neurol Neurosurg Psychiatry (1990) 53(3):200–7. doi:10.1136/jnnp.53.3.200
CrossRef Full Text | Google Scholar
15. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology (2006) 66(10):1485–9. doi:10.1212/01.wnl.0000216139.44259.74
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology (2015) 85:177–89. doi:10.1212/WNL.0000000000001729
CrossRef Full Text | Google Scholar
17. Kira J. The expanding phenotype of CLIPPERS: is it a disease or a syndrome? J Neurol Neurosurg Psychiatry (2012) 83(1):2–3. doi:10.1136/jnnp-2011-301626
CrossRef Full Text | Google Scholar
18. Henn V, Lang W, Hepp K, Reisine H. Experimental gaze palsies in monkeys and their relation to human pathology. Brain (1984) 107(2):619–36. doi:10.1093/brain/107.2.619
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Büttner-Ennever JA, Buttner U. A cell group associated with vertical eye movements in the rostral mesencephalic reticular formation of the monkey. Brain Res (1978) 151(1):31–47. doi:10.1016/0006-8993(78)90948-4
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Horn AKE. The reticular formation. Prog Brain Res (2006) 151:127–55. doi:10.1016/S0079-6123(05)51005-7
CrossRef Full Text | Google Scholar
21. Nakao S, Shiraishi Y, Oda H, Inagaki M. Direct inhibitory projection of pontine omnipause neurons to burst neurons in the Forel’s field H controlling vertical eye movement-related motoneurons in the cat. Exp Brain Res (1988) 70(3):632–6. doi:10.1007
CrossRef Full Text | Google Scholar
22. Bender MB. Brain control of conjugate horizontal and vertical eye movements: a survey of the structural and functional correlates. Brain (1980) 103(1):23–69. doi:10.1093/brain/103.1.23
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Ranalli PJ, Sharpe JA. Vertical vestibulo-ocular reflex, smooth pursuit and eye-head tracking dysfunction in internuclear ophthalmoplegia. Brain (1988) 111(Pt 6):1299–317.
PubMed Abstract | Google Scholar
24. Bhidayasiri R, Plant GT, Leigh RJ. A hypothetical scheme for the brainstem control of vertical gaze. Neurology (2000) 54(10):1985–93.
PubMed Abstract | Google Scholar