Neutropenia
Lista wszystkich potencjalnych przyczyn neutropenii nie jest krótka. Etiologię neutropenii można rozpatrywać na dwa szerokie sposoby, według mechanizmu lub kategorii etiologicznej.
Mechanizmy wywołujące neutropenię są zróżnicowane i nie do końca poznane. W wielu przypadkach neutropenia występuje po długotrwałej ekspozycji na lek lub inną substancję, co powoduje zmniejszenie produkcji neutrofilów przez hipoplastyczny szpik kostny. Sugeruje to bezpośrednie działanie toksyczne na komórki macierzyste. W innych przypadkach konieczne jest powtarzające się, ale przerywane narażenie na lek lub inną substancję. Sugeruje to mechanizm immunologiczny, chociaż ta koncepcja nie została udowodniona. W wielu sytuacjach klinicznych dokładna ekspozycja i czas jej trwania w odniesieniu do początku neutropenii nie są znane.
Wobec niepełnego zrozumienia mechanizmów neutropenii łatwiejsza do utrzymania jest klasyfikacja według szerokiej kategorii etiologicznej. W tym schemacie etiologia neutropenii może być sklasyfikowana jako wrodzona (dziedziczna) lub nabyta. Chociaż ta kategoryzacja może mieć ograniczoną przydatność diagnostyczną, może być przydatna do wyraźnego oddzielenia dziedzicznych przyczyn neutropenii od całej gamy przyczyn nabytych. W przypadku neutropenii dziedzicznych zaburzenia te mogą być dalej opisywane jako związane z izolowaną neutropenią lub z innymi defektami, zarówno immunologicznymi, jak i fenotypowymi.
Wiele zaburzeń dziedzicznych jest spowodowanych mutacjami w genie kodującym elastazę neutrofilową, ELA2. W grę wchodzi kilka alleli. Najczęstsze mutacje to substytucje intronowe, które inaktywują miejsce splicingu w intronie 4. Zaangażowane są również inne geny niż ELA2. W tabeli poniżej wymieniono niektóre z występujących schorzeń genetycznych; są to schorzenia rzadkie.
Tabela 1. Genetyczne (dziedziczne) uwarunkowania w agranulocytozie (Otwórz tabelę w nowym oknie)
|
Syndrom |
Inheritance |
Gene |
Clinical Features |
|
Cyclic neutropenia |
Autosomal dominant |
ELA2 |
Alternate 21-day cycling of neutrophils and monocytes |
|
Kostmann syndrome |
Autosomal recessive |
Unknown |
Stable neutropenia, no MDS or AML |
|
Severe congenital neutropenia |
Autosomal dominant |
ELA2 (35-84%) |
Stable neutropenia, MDS or AML |
|
Autosomal dominant |
GFI1 |
Stable neutropenia, circulating myeloid progenitors, lymphopenia |
|
|
Sex linked |
Wasp |
Neutropenic variant of Wiskott-Aldrich syndrome |
|
|
Autosomal dominant |
G-CSFR |
G-CSF–refractory neutropenia, no AML or MDS |
|
|
Hermansky-Pudlak syndrome type 2 |
Autosomal recessive |
AP3B1 |
Severe congenital neutropenia, platelet dense-body defect, oculocutaneous albinism |
|
Chediak-Higashi syndrome |
Autosomal recessive |
LYST |
Neutropenia, oculocutaneous albinism, giant lysosomes, impaired platelet function |
|
Barth syndrome |
Sex linked |
TAZ |
Neutropenia, often cyclic; cardiomyopathy, methylglutaconic aciduria |
|
Cohen syndrome |
Autosomal recessive |
COH1 |
Neutropenia, mental retardation, dysmorphism |
|
Source: Modified from Berliner et al, 2004. AML = acute myeloid leukemia; G-CSF = granulocyte colony-stimulating factor; MDS = myelodysplastic syndrome. |
|||
Przyczyny neutropenii nabytej są złożone, ale większość z nich jest związana z trzema głównymi kategoriami: zakażeniem, lekami (zarówno bezpośrednio toksycznymi, jak i pośredniczącymi w układzie immunologicznym) oraz autoagresją. Przewlekła łagodna neutropenia, lub przewlekła neutropenia idiopatyczna, wydaje się być zaburzeniem nakładającym się z formami dziedzicznymi i nabytymi, i czasami jest nie do odróżnienia. Niektórzy pacjenci z neutropenią podają jasny wywiad i wzór rodzinny, podczas gdy inni nie mają wywiadu rodzinnego, mają niewiele oznaczeń w badaniach krwi i nieznany czas trwania neutropenii. Ta grupa pacjentów może mieć neutropenię dziedziczną lub nabytą. Poniżej przedstawiono krótkie podsumowanie wrodzonych i nabytych zaburzeń neutropenicznych.
Wrodzona neutropenia z towarzyszącymi defektami immunologicznymi
Neutropenię z nieprawidłowymi immunoglobulinami obserwuje się u osób z agammaglobulinemią sprzężoną z chromosomem X, izolowanym niedoborem immunoglobuliny A (IgA), zespołem hiperimmunoglobuliny M sprzężonej z chromosomem X (XHIGM) i dysgammaglobulinemią typu I. W XHIGM, który jest spowodowany mutacjami w ligandzie CD40, pacjenci mogą mieć prawidłowy lub podwyższony poziom IgM, ale znacznie obniżony poziom IgG w surowicy. We wszystkich tych zaburzeniach ryzyko infekcji jest wysokie, a leczenie polega na podawaniu dożylnych immunoglobulin (IVIG).
Pacjenci z dysgenezją siatkówki wykazują ciężką neutropenię, brak odporności komórkowej, agammaglobulinemię i limfopenię. Występują zagrażające życiu infekcje, które są oporne na działanie czynnika stymulującego tworzenie kolonii granulocytów (G-CSF). Przeszczep szpiku kostnego jest leczeniem z wyboru.
Wrodzona lub przewlekła neutropenia
Szeroko wrodzona neutropenia (SCN), lub zespół Kostmanna, jest najczęściej spowodowana dziedziczeniem recesywnym i występuje w odległych, izolowanych populacjach o wysokim stopniu pokrewieństwa. Opisywano również przypadki autosomalne dominujące i sporadyczne, najczęściej spowodowane mutacjami w receptorze G-CSF. W zespole tym nie ma jednolitego defektu genetycznego. Mutacje w ELA2, które są przyczyną cyklicznej neutropenii (patrz niżej) nie są wystarczające do wyjaśnienia fenotypu Kostmann-like SCN.
Pacjenci pojawiają się w wieku 3 miesięcy z nawracającymi infekcjami bakteryjnymi. Najczęstszym miejscem zakażenia jest jama ustna i okolica odbytu. Ten typ neutropenii jest ciężki, a leczenie polega na podawaniu G-CSF. Ryzyko konwersji do zespołu mielodysplastycznego (MDS)/ostrej białaczki szpikowej (AML) z monosomią 7 po leczeniu G-CSF jest związane z dodatkowymi nabytymi mutacjami. Większość z tych przypadków jest spowodowana mutacją w receptorze G-CSF. Pacjenci, u których występuje kliniczna odpowiedź na G-CSF, są leczeni dożywotnio.
Niektórzy pacjenci z innymi postaciami SCN wydają się mieć mutacje w GFI1, genie represora transkrypcji z palcem cynkowym, zaangażowanym w funkcję krwiotwórczych komórek macierzystych i decyzje o wyborze linii.
Cykliczna neutropenia (CN) charakteryzuje się okresowymi wybuchami neutropenii związanej z infekcją, po których następuje odbudowa liczby neutrofilów obwodowych. Jej okresowość wynosi około 21 dni (zakres, 12-35 dni). Prekursory granulocytów znikają ze szpiku przed każdym nadirem neutrofilów w cyklu z powodu przyspieszonej apoptozy mieloidalnych komórek progenitorowych. Niektóre przypadki mogą być uwarunkowane genetycznie z dziedziczeniem autosomalnym recesywnym. Inne przypadki mogą być spowodowane dziedziczeniem autosomalnym dominującym. W niektórych sporadycznych przypadkach CN, pacjenci mają mutacje w ELA2.
Osoby z CN zazwyczaj występują jako niemowlęta lub dzieci, ale istnieją nabyte formy CN w wieku dorosłym. Rokowanie jest dobre, przebieg łagodny, jednak u 10% pacjentów wystąpią zagrażające życiu infekcje. Leczenie cyklicznej neutropenii polega na codziennym podawaniu G-CSF.
Przewlekła łagodna neutropenia
Rodzinna przewlekła łagodna neutropenia, lub łagodna neutropenia etniczna, jest zaburzeniem z autosomalnym dominującym wzorcem dziedziczenia obserwowanym w afrykańskim, jemeńskim żydowskim, etiopskim żydowskim, arabskim, karaibskim i zachodnioindyjskim pochodzeniu. W populacjach pochodzenia afrykańskiego i jemeńskiego badania genetyczne wykazują silny związek z polimorfizmem pojedynczego nukleotydu w genie DARC. Pacjenci są zazwyczaj bezobjawowi, a infekcje mają łagodny przebieg. U osób z przewlekłą łagodną neutropenią ryzyko wystąpienia infekcji jest niskie i nie jest wymagana żadna specyficzna terapia.
W nierodzinnej przewlekłej łagodnej neutropenii, łagodne infekcje o łagodnym przebiegu są typowe dla tego zaburzenia. ANC reaguje jednak na stres, taki jak infekcje, kortykosteroidy i katecholaminy.
Idiopatyczna przewlekła ciężka neutropenia
Idiopatyczna przewlekła ciężka neutropenia jest rozpoznaniem z wykluczenia. U pacjentów dotkniętych chorobą występują infekcje i ciężka neutropenia.
Neutropenia związana z nieprawidłowościami fenotypowymi
Zespół Shwachmana (Shwachman-Diamond) ma autosomalny recesywny sposób dziedziczenia. Neutropenia jest umiarkowana do ciężkiej, ze śmiertelnością 15-25%, a zespół objawia się w okresie niemowlęcym, nawracającymi infekcjami, biegunką i trudnościami w karmieniu. Może wystąpić karłowatość, chondrodysplazja i niewydolność zewnątrzwydzielnicza trzustki.
Zespół Shwachmana-Diamonda i sprzężona z chromosomem X dyskeratoza wrodzona (DC), hipoplazja chrzęstno-włosowa (CHH) i niedokrwistość Diamonda-Blackfana (DBA) wydają się mieć wspólne defekty genów zaangażowanych w syntezę rybosomów. Większość przypadków zespołu Shwachmana-Diamonda jest spowodowana mutacjami w genie SBDS. Dokładna funkcja tego genu jest wciąż wyjaśniana, jednak jest on zaangażowany w syntezę rybosomów i reakcje przetwarzania RNA. Leczenie polega na podawaniu G-CSF.
W CHH wzór dziedziczenia jest autosomalny recesywny na chromosomie 9 i występuje w rodzinach amiszów i Finów. CHH jest spowodowana mutacjami w genie RMRP, który koduje składnik RNA kompleksu RNazy MRP (ribonuclease mitochondrial RNA processing). Neutropenia jest umiarkowana do ciężkiej. CHH objawia się defektami odporności komórkowej, niedokrwistością makrocytarną, chorobami przewodu pokarmowego i karłowatością. Wykazuje również predyspozycje do chorób nowotworowych, zwłaszcza chłoniaków. Leczenie polega na przeszczepie szpiku kostnego.
Dyskeratosis congenita (zespół Zinssera-Cole-Engmana) objawia się opóźnieniem umysłowym, pancytopenią i defektem odporności komórkowej. Dyskeratoza wrodzona występuje częściej u mężczyzn niż u kobiet i jest hematologicznie podobna do niedokrwistości Fanconiego. Dyskeratoza wrodzona jest zwykle recesywna sprzężona z chromosomem X, chociaż istnieją formy autosomalne dominujące i autosomalne recesywne tego zaburzenia.
Recesywna forma zaburzenia sprzężona z chromosomem X została powiązana z mutacjami w DKC1, która koduje dyskerynę, białko nukleolarne związane z cząsteczkami rybonukleoprotein. Postać autosomalna dominująca związana jest z mutacjami w innym genie, TERC, który jest częścią telomerazy. Telomeraza ma zarówno komponent białkowy, jak i RNA, a TERC koduje komponent RNA. Pacjenci z tym zaburzeniem mają krótsze telomery niż normalnie. Leczenie polega na podawaniu G-CSF, czynnika stymulującego kolonie granulocytów-makrofagów (GM-CSF) i przeszczepie szpiku kostnego.
Zespół Bartha jest zaburzeniem recesywnym sprzężonym z chromosomem X, objawiającym się kardiomiopatią w okresie niemowlęcym, miopatią szkieletową, nawracającymi infekcjami, karłowatością i umiarkowaną do ciężkiej neutropenią.
Zespół Chediak-Higashi jest zaburzeniem dziedziczonym autosomalnie recesywnie, charakteryzującym się nawracającymi infekcjami, spowolnieniem umysłowym, światłowstrętem, oczopląsem, albinizmem okularowym, neuropatią, zaburzeniami krwawienia, zapaleniem dziąseł i obecnością ziarnistości lizosomalnych w różnych komórkach. Neutropenia jest umiarkowana do ciężkiej, a leczenie polega na przeszczepie szpiku kostnego.
Myelokathexis
Myelokathexis występuje w okresie niemowlęcym z umiarkowaną neutropenią i wiąże się z nawracającymi infekcjami. Stan ten jest spowodowany przyspieszoną apoptozą i obniżoną ekspresją bcl-x w prekursorach neutrofilów. Obserwuje się nieprawidłowy wygląd jądra, z hipersegmentacją z pasmami jądrowymi, pyknozą i cytoplazmatyczną wakuolizacją. W leczeniu stosuje się G-CSF i GM-CSF.
Zespół leniwych leukocytów
Zespół leniwych leukocytów jest ciężką neutropenią z towarzyszącą nieprawidłową ruchliwością neutrofilów. Etiologia jest nieznana, a leczenie ma charakter wspomagający.
Zaburzenia metaboliczne
Są to przewlekłe neutropenie ze zmienną wartością ANC. They include glycogen storage disease type 1b and various acidemias, such as isovaleric, propionic, and methylmalonic. In glycogen storage disease type 1b, the treatment is G-CSF and GM-CSF.
Acquired neutropenia caused by intrinsic bone marrow disease
Intrinsic bone marrow diseases that may cause neutropenia include the following:
-
Aplastic anemia
-
Hematologic malignancy (eg, leukemia, lymphoma, myelodysplasia, myeloma)
-
Ionizing radiation
-
Tumor infiltration
-
Granulomatous infection
-
Myelofibrosis
Immune-mediated neutropenia
A drug may act as a hapten and induce antibody formation. This mechanism operates in cases due to gold, aminopyrine, and antithyroid drugs. The antibodies destroy the granulocytes and may not require the continued presence of the drug for their action. Alternatively, the drug may form immune complexes that attach to the neutrophils. This mechanism operates with quinidine.
Drug immune-mediated neutropenia may be caused by the following:
-
Aminopyrine
-
Quinidine
-
Cephalosporins
-
Penicillins
-
Sulfonamides
-
Phenothiazines
-
Hydralazine
Other medications have been implicated
Autoimmune neutropenia is the neutrophil analogue of autoimmune hemolytic anemia and of idiopathic thrombocytopenic neutropenia. It should be considered in the absence of any of the common causes. Antineutrophil antibodies have been demonstrated in these patients. Autoimmune neutropenia may be associated with the following:
-
Rheumatoid arthritis (with or without Felty syndrome)
-
Sjögren syndrome
-
Chronic, autoimmune hepatitis
-
Systemic lupus erythematosus
-
Thymoma
-
Goodpasture disease
-
Granulomatosis with polyangiitis (Wegener granulomatosis)
-
Pure red blood cell (RBC) aplasia, in which there is complete disappearance of granulocyte tissue from the bone marrow; pure RBC dysplasia is a rare disorder due to the presence of antibody-mediated, granulocyte-macrophage colony forming unit (GM-CFU) inhibitory activity, and it is often associated with thymoma
-
Transfusion reactions, which can be caused by the surface antigens of neutrophilia;
-
Rozrost dużych ziarnistych limfocytów lub białaczka
W izoimmunizacyjnej neutropenii noworodków matka wytwarza przeciwciała IgG przeciwko neutrofilom do antygenów neutrofilów płodu, które są rozpoznawane jako nie-ja. Zdarza się to u 3% żywych urodzeń. Zaburzenie objawia się gorączką noworodkową, zakażeniem dróg moczowych, cellulitisem, zapaleniem płuc i sepsą. Czas trwania neutropenii wynosi zwykle 7 tygodni.
Przewlekła neutropenia autoimmunologiczna obserwowana jest u osób dorosłych i nie wykazuje predylekcji wiekowej. Aż u 36% pacjentów stwierdza się obecność przeciwciał przeciwneutrofilowych w surowicy, a przebieg kliniczny jest zwykle mniej ciężki. Pacjenci mogą mieć to zaburzenie w skojarzeniu z toczniem rumieniowatym układowym, reumatoidalnym zapaleniem stawów, ziarniniakiem Wegenera, przewlekłym zapaleniem wątroby.
Jeżeli przewlekła neutropenia autoimmunologiczna jest związana z tymi chorobami, w leczeniu wskazane jest stosowanie kortykosteroidów. U noworodków i dzieci zaburzenie to wiąże się z mniejszym ryzykiem infekcji i łagodniejszymi zakażeniami obejmującymi ucho środkowe, przewód pokarmowy i skórę.
Limfocytoza T-gamma, czyli zaburzenie limfoproliferacyjne, jest chorobą klonalną limfocytów T CD3+ lub komórek NK (natural killer) CD3-, które naciekają szpik kostny i tkanki. Znana również jako białaczka dużych ziarnistych limfocytów (białaczka LGL), limfocytoza T-gamma może być związana z reumatoidalnym zapaleniem stawów i jest związana z wysokim mianem przeciwciał antyneutrofilowych. Neutropenia jest uporczywa i ciężka. Leczenie ma często charakter podtrzymujący, ale jest również ukierunkowane na eliminację populacji klonalnej.
Nabyta neutropenia spowodowana zakażeniem
Zakażenia są najczęstszą formą nabytej neutropenii. Zakażenia, które mogą powodować neutropenię obejmują, ale nie są ograniczone do następujących:
-
Bacterial sepsis
-
Viral infections (eg, influenza, measles, Epstein Barr virus , cytomegalovirus , viral hepatitis, human immunodeficiency virus -1) (see first image below)
-
Toxoplasmosis
-
Brucellosis
-
Typhoid
-
Tuberculosis (see second and third images below)
-
Malaria
-
Dengue fever
-
Rickettsial infection
-
Babesiosis
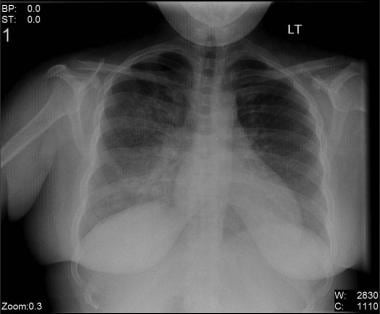 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia. 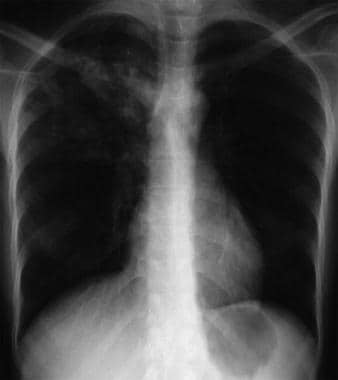 Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Kobieta została przyjęta do izolatki i w warunkach ambulatoryjnych rozpoczęła leczenie empiryczne 4 lekami. Gruźlicę potwierdzono badaniem plwociny. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Kobieta została przyjęta do izolatki i w warunkach ambulatoryjnych rozpoczęła leczenie empiryczne 4 lekami. Gruźlicę potwierdzono badaniem plwociny. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org. 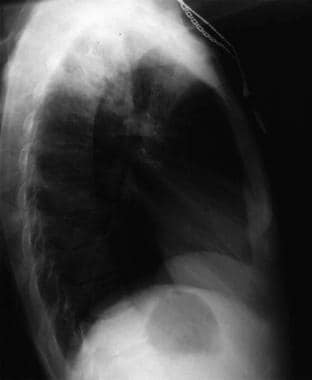 Boczny radiogram klatki piersiowej u 31-letniego pacjenta z grypowym zapaleniem płuc. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org.
Boczny radiogram klatki piersiowej u 31-letniego pacjenta z grypowym zapaleniem płuc. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org.
 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.  Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Kobieta została przyjęta do izolatki i w warunkach ambulatoryjnych rozpoczęła leczenie empiryczne 4 lekami. Gruźlicę potwierdzono badaniem plwociny. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Kobieta została przyjęta do izolatki i w warunkach ambulatoryjnych rozpoczęła leczenie empiryczne 4 lekami. Gruźlicę potwierdzono badaniem plwociny. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org.  Boczny radiogram klatki piersiowej u 31-letniego pacjenta z grypowym zapaleniem płuc. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org.
Boczny radiogram klatki piersiowej u 31-letniego pacjenta z grypowym zapaleniem płuc. Obraz dzięki uprzejmości Remote Medicine, remotemedicine.org. Najczęściej zakażone organizmy pochodzą z flory endogennej. Organizmy Staphylococcus aureus występują w przypadkach zakażeń skóry. Organizmy Gram-ujemne obserwuje się w zakażeniach układu moczowego i pokarmowego, szczególnie Escherichia coli i Pseudomonas species. Mogą również wystąpić zakażenia Candida albicans. W jamie ustnej może występować flora mieszana.
Zakażenia wirusowe często prowadzą do łagodnej lub umiarkowanej neutropenii. Agranulocytoza jest rzadka, ale może wystąpić. Najczęstsze organizmy to wirus Epsteina-Barr, wirus zapalenia wątroby typu B, wirus żółtej gorączki, wirus cytomegalii i grypy. Wiele przytłaczających zakażeń, zarówno wirusowych, jak i bakteryjnych, może powodować ciężką neutropenię.
Nabyta neutropenia spowodowana niedoborami żywieniowymi
Zaburzenia żywieniowe, które mogą powodować neutropenię, obejmują niedobór witaminy B-12, folianów i miedzi.
Nabyta neutropenia spowodowana przez leki i substancje chemiczne, z wyłączeniem chemioterapii cytotoksycznej
Liczna liczba leków została powiązana z neutropenią. Do kategorii największego ryzyka należą leki przeciwtarczycowe, makrolidy i prokainamidy. Jak wspomniano powyżej, wiele leków działa poprzez mechanizm immunologiczny. Jednakże, niektóre leki wydają się mieć bezpośredni toksyczny wpływ na komórki macierzyste szpiku lub prekursory neutrofilów w przedziale mitotycznym. Na przykład, leki takie jak leki przeciwpsychotyczne i przeciwdepresyjne oraz chloramfenikol mogą działać jako bezpośrednie toksyny u niektórych osób, w oparciu o metabolizm i wrażliwość w ten sposób. Inne leki mogą mieć kombinację mechanizmów immunologicznych i nieimmunologicznych lub mogą mieć nieznane mechanizmy działania.
Środki przeciwdrobnoustrojowe obejmują penicylinę, cefalosporyny, wankomycynę, chloramfenikol, gentamycynę, klindamycynę, doksycyklinę, flucytozynę, nitrofurantoinę, nowobiocynę, minocyklinę, gryzeofulwinę, linkomycynę, metronidazol, ryfampinę, izoniazyd, streptomycynę, tiacetazon, mebendazol, pirymetamina, lewamizol, ristocetyna, sulfonamidy, chlorochina, hydroksychlorochina, chinakryna, etambutol, dapson, cyprofloksacyna, trimetoprim, imipenem/kilastatyna, zidowudyna, fludarabina, acyklowir i terbinafina.
Środki przeciwbólowe i przeciwzapalne obejmują aminopirynę, dipyron, indometacynę, ibuprofen, kwas acetylosalicylowy, diflunisal, sulindak, tolmetynę, benoksaprofen, barbiturany, mesalazynę i chininę.
Antypsychotyki, leki przeciwdepresyjne i środki neurofarmakologiczne obejmują fenotiazyny (chlorpromazyna, metylpromazyna, mepazyna, promazyna, tiorydazyna, prochlorperazyna, trifluoperazyna, trimeprazyna), klozapina, risperidon, imipramina, desipramina, diazepam, chlordiazepoksyd, amoksapina, meprobamat, tiotyksen i haloperidol.
Leki przeciwdrgawkowe obejmują kwas walproinowy, fenytoinę, trimetadion, mefenytoinę (Mesantoin), etosuksymid i karbamazepinę.
Leki przeciwtarczycowe obejmują tiouracyl, propylotiouracyl, metimazol, karbimazol, nadchloran potasu i tiocyjanian.
Leki sercowo-naczyniowe obejmują prokainamid, kaptopril, aprydynę, propranolol, hydralazynę, metyldopę, chinidynę, diazoksyd, nifedypinę, propafenon, tiklopidynę i vesnarinon.
Leki przeciwhistaminowe obejmują cymetydynę, ranitydynę, tripelenaminę (piribenzaminę), metafenilen, tenalidynę, bromfenidynę i mianserynę.
Leki moczopędne obejmują acetazolamid, bumetanid, chlorotiazyd, hydrochlorotiazyd, chlortalidon, metazolamid i spironolakton.
Środki hipoglikemizujące obejmują chlorpropamid i tolbutamid.
Leki przeciwmalaryczne obejmują amodiaquine, dapsone, hydroxychloroquine, pyrimethamine, and quinine.
Leki różne obejmują allopurinol, kolchicynę, aminoglutetymid, famotydynę, bezafibrat, flutamid, tamoksyfen, penicylaminę, kwas retinowy, metoklopramid, fenindion, dinitrofenol, kwas etakrynowy, dichlorodifenylotrichloroetan (DDT), cynchofen, antymon, pirytilodion, rauwolfia, etanol, chlorpropamid, tolbutamid, tiazydy, spironolakton, metazolamid, acetazolamid, IVIG i lewodopa.
Metale ciężkie obejmują złoto, arsen i rtęć.
Narażenie na leki lub substancje chemiczne jest najczęstszą przyczyną agranulocytozy: około połowa pacjentów ma historię narażenia na leki lub substancje chemiczne. Każda substancja chemiczna lub lek, który może obniżyć szpik kostny i spowodować hipoplazję lub aplazję jest w stanie wywołać agranulocytozę. Niektóre leki robią to u wszystkich, jeśli są podawane w wystarczająco dużych dawkach. Inne środki wydają się powodować idiosynkratyczne reakcje, które dotyczą tylko niektórych podatnych osób.
Niektóre środki (np. kwas walproinowy, karbamazepina i antybiotyki beta-laktamowe) działają poprzez bezpośrednie hamowanie mielopoezy. W hodowlach szpiku kostnego, środki te hamują tworzenie kolonii granulocytów w sposób zależny od dawki. W większości pozostałych przypadków rolę odgrywa bezpośrednie uszkodzenie mikrośrodowiska szpiku kostnego lub prekursorów mieloidalnych.
Wiele leków związanych z agranulocytozą zostało zgłoszonych do Amerykańskiej Agencji Żywności i Leków (FDA) w ramach obowiązku zgłaszania działań niepożądanych. Wiele leków zostało również zgłoszonych do rejestru prowadzonego przez American Medical Association (AMA). Zgłoszone leki były stosowane samodzielnie, w połączeniu z innym lekiem, o którym wiadomo, że jest potencjalnie toksyczny, lub z innym lekiem bez znanej toksyczności. Kilka leków jest szczególnie wartych uwagi ze względu na ich wysoką częstość powiązań z agranulocytozą. Należą do nich następujące leki:
-
Phenothiazine
-
Antithyroid drugs (thiouracil and propylthiouracil)
-
Aminopyrine
-
Chloramphenicol
-
Sulfonamides
Miscellaneous immunologic neutropenias
Immunologic neutropenias may occur after bone marrow transplantation and blood product transfusions.
Felty syndrome is a syndrome of rheumatoid arthritis, splenomegaly, and neutropenia. Splenectomy shows an initial response, but neutropenia may recur in 10-20% of patients. Treatment is directed toward rheumatoid arthritis.
In complement activation–mediated neutropenia, hemodialysis, cardiopulmonary bypass, and extracorporeal membrane oxygenation (ECMO) expose blood to artificial membranes and can cause complement activation with subsequent neutropenia.
In splenic sequestration, the degree of neutropenia resulting from this process is proportional to the severity of the splenomegaly and the bone marrow’s ability to compensate for the reduction in circulating bands and neutrophils.
Eosinopenia and basophilopenia
Eosinopenia may be associated with the following:
-
Acute bacterial infection
-
Glucocorticoid administration
-
Physical stress
-
Thymoma
Decreased circulating basophils may be associated with the following:
-
Anaphylaxis
-
Acute infection
-
Drug-induced hypersensitivity
-
Congenital absence of basophils
-
Hemorrhage
-
Hyperthyroidism
-
Ionizing radiation
-
Neoplasia
-
Ovulation
-
Urticaria
-
Drugs (eg, corticosteroid, adrenocorticotropic hormone therapy, chemotherapeutic agents, thyroid hormones)
Go to Pediatric Autoimmune and Chronic Benign Neutropenia for complete information on this topic.