Określenie mutacji V617F genu JAK2 w przewlekłych zespołach mieloproliferacyjnych w naszym kraju: opis przypadku
Określenie mutacji V617F genu JAK2 w przewlekłych zespołach mieloproliferacyjnych w naszym kraju: opis przypadku
Dr Daniela Lens*, Pablo Muxi†, Lics. Andreína Brugnini‡,
Natalia Trías‡, Dra. Silvia Pierri§
Hospital de Clínicas. Wydział Lekarski.
Uniwersytet Republiki. Montevideo, Urugwaj
Abstrakt
Polycythemia vera (PV), małopłytkowość istotna (ET) i idiopatyczna mielofibroza (IM) są blisko spokrewnionymi klonalnymi chorobami mieloproliferacyjnymi charakteryzującymi się nadmierną proliferacją jednej lub więcej linii mieloidalnych, takich jak erytrocyty, płytki krwi i fibroblasty w szpiku kostnym.
Pomimo że istnieją ścisłe kryteria rozpoznania tych zespołów mieloproliferacyjnych, dokładna kategoryzacja pozostaje kwestią dyskusyjną, a dodatkowo zaburzenia te są w wielu przypadkach trudne do odróżnienia od procesów reaktywnych.
Ostatnio, w 2005 roku, w kilku z tych jednostek zidentyfikowano mutację w genie kinazy tyrozynowej Janus kinase 2 (JAK2). Mutacja ta polega na zamianie G na T w pozycji 1849, co skutkuje zamianą fenyloalaniny na walinę w białku (JAK2 V617F).
Częstość występowania tej mutacji stwierdzono w około 90% przypadków z PV i w około 50% przypadków z IM i ET.
W niniejszej pracy opiszemy wykrycie tej mutacji u pacjenta z rozpoznaniem prawdopodobnej PV przy użyciu wysoce czułego, specyficznego dla alleli testu łańcuchowej reakcji polimerazy (PCR) do wykrywania tej mutacji oraz omówimy znaczenie tej niedawno odkrytej mutacji w diagnostyce i leczeniu zespołów mieloproliferacyjnych BCR-ABL-ujemnych.
Słowa kluczowe: MYELOPROLIFERATIVE DISORDERS – diagnosis.
MUTACJA – genetyka.
PROTEIN-TYROSINE KINASE.
* Profesor nadzwyczajny. Podstawowy Wydział Lekarski.
† Profesor nadzwyczajny. Klinika Hematologii. Kliniczny Wydział Lekarski.
‡ Biochemia Lic. Asystent. Podstawowy Wydział Lekarski.
§ Profesor nadzwyczajny. Klinika Hematologii. Wydział Kliniczny Medycyny.
Korespondencja: Dr. Daniela Lens.
Dział. Medycyna podstawowa. Hospital de Clínicas. Piso 15. Avda Italia s/n. Montevideo CP 11600. Montevideo, Urugwaj.
Email: [email protected]
Przyjęto: 31/7/06.
Zaakceptowano: 26/2/07.
Wprowadzenie
W przeciwieństwie do przewlekłej białaczki szpikowej, w której identyfikacja specyficznej rearanżacji BCR-ABL doprowadziła do przełomu w diagnostyce, monitorowaniu i leczeniu tej jednostki chorobowej, W przewlekłych zespołach mieloproliferacyjnych (PMS) BCR-ABL ujemnych, takich jak polytemia vera (PV), małopłytkowość istotna (ET) i idiopatyczna mielofibroza (IM), do niedawna nie znano specyficznych zmian genetycznych.
Między majem a czerwcem 2005 roku, pięć grup badaczy opisało nową mutację punktową w genie kinazy tyrozynowej JAK2, spowodowaną zamianą guaniny na tyminę w nukleotydzie 1849 w eksonie 14, co skutkuje zamianą waliny na fenyloalaninę w pozycji 617 białka, dla której mutacja JAK2 została nazwana V617F(1-5).
Mutacja ta była obserwowana w około 90% przypadków PV, gdy do jej wykrycia używano testów o wysokiej czułości(6). Stwierdzono ją również w około 50% przypadków IM i ET, natomiast nie znaleziono jej u osób zdrowych ani u pacjentów z wtórną erytrocytozą, tak więc mutacja ta ma bardzo wysoką wartość predykcyjną w odróżnianiu SMPC od stanów nieklonalnych, takich jak wtórna policytemia.
Mutacja ta występuje w wysoce konserwowanym regionie autoinhibicyjnej domeny, która negatywnie reguluje sygnalizację JAK2. Wiele badań wykazało, że mutacja ta jest zaangażowana w patogenezę tych schorzeń, szczególnie ze względu na zysk funkcji genu JAK2 oraz poprzez utratę kontroli, które są związane z nadmierną mieloproliferacją w tych zaburzeniach.
W niniejszym artykule opiszemy wykrycie tej mutacji u pacjenta z rozpoznaniem prawdopodobnej PV i omówimy znaczenie tej nowej mutacji w diagnostyce i leczeniu BCR-ABL ujemnych SMP.
Materiał i metoda
Ekstrakcja kwasu dezoksyrybonukleinowego
Ekstrakcja genomowego kwasu dezoksyrybonukleinowego (DNA) została przeprowadzona z 1 ml cytrynianowej krwi obwodowej, przy użyciu komercyjnego odczynnika dnazol (Life Techno-logies) i zgodnie z protokołem wskazanym przez producenta. Próbki DNA przechowywano w temperaturze 4°C.
Wykrywanie mutacji Vl617F
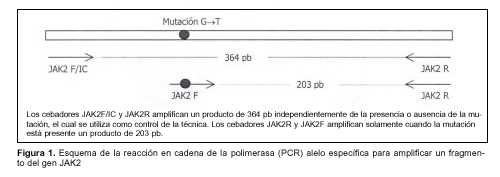
Mutację V617F wykrywano z uprzednio uzyskanego genomowego DNA za pomocą specyficznej dla alleli reakcji łańcuchowej polimerazy (PCR) zgodnie z metodą opublikowaną przez Baxtera i wsp.(1). Ten PCR został zaprojektowany do użycia trzech różnych starterów, zwanych JAK2R, JAK2F/IC i JAK2F (tabela 1). Starter JAK2F jest specyficzny dla zmutowanego allelu, a starter JAK2F/IC występuje u wszystkich osobników niezależnie od obecności lub braku mutacji, stanowiąc tym samym wewnętrzną kontrolę reakcji. I tak, u wszystkich osób przy użyciu starterów JAK2F/IC i JAK2R amplifikuje się produkt o długości 364 par zasad (bp), a tylko u osób z badaną mutacją uzyskuje się produkt o długości 203 bp przy użyciu starterów JAK2F i JAK2R (ryc. 1).
PCR amplifikację przeprowadzono w końcowej objętości 20 uL używając 500 ng DNA, 200 mM/L każdego z dNTPs, 0,5 U polimerazy Taq (New England Biolabs) i 2 uL 10x buforu Taq zawierającego 2 mM MgCl (New England Biolabs), 0,5 mM starterów JAK2 F i JAK2 F/IC oraz 1 mM startera JAK2 R.
Użyty program PCR składa się z 36 cykli, rozpoczynających się od 5-minutowej denaturacji, następnie cykli: 30 sekund w 94°C, 30 sekund w 58°C i 30 sekund w 72°C, kończących się końcowym 5-minutowym przedłużeniem. Kontrola negatywna zawierająca wszystkie odczynniki niezbędne do amplifikacji, w której szablon DNA jest zastąpiony wodą, była zawsze włączona do wszystkich reakcji. Weryfikację oczekiwanego produktu PCR przeprowadzono na 2% żelu agarozowym barwionym bromkiem etydyny(7).

Analiza przypadku
Mężczyzna w wieku 72 lat, nałogowy palacz tytoniu, z przewlekłym nadciśnieniem tętniczym, leczony, nosiciel przewlekłej arteriopatii obturacyjnej, u którego w 2002 roku wykonano amputację piątego palca stopy lewej. W grudniu 2004 roku, po wystąpieniu nowego epizodu obturacyjnego w lewej kończynie dolnej, w hematogramie stwierdzono wzrost liczby krwinek czerwonych z hematokrytem 66%, granulocytozą 24 000 i liczbą płytek krwi 245 000. Hematokryt pozostał bez zmian, a liczba płytek krwi stopniowo wzrosła do 710 000. W badaniu biopsyjnym szpiku kostnego stwierdzono hiperkomórkowy szpik kostny z hiperplazją szeregu erytroidalnego, co pozwoliło wnioskować o przewlekłym zespole mieloproliferacyjnym typu PV.
Pacjentka była leczona flebotomiami, a w maju 2005 roku rozpoczęła leczenie hydroksymocznikiem, które przerwano w lutym 2006 roku z powodu powikłania krwotocznego.
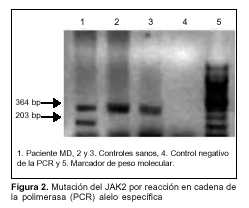
Jak przedstawiono na rycinie 2, pacjentka wykazywała wzór z dwoma pasmami w badaniu PCR specyficznym dla alleli mutacji V617F genu JAK2. Z jednej strony, pasmo kontrolne 364 bp, które jest również obecne w dwóch badanych zdrowych grupach kontrolnych. Z drugiej strony, u pacjenta amplifikowano również produkt 203 bp wskazujący na obecność mutacji. Pasma tego nie obserwowano w zdrowych grupach kontrolnych.
Dyskusja i wnioski
W niniejszej pracy opisaliśmy pierwszy w naszym kraju przypadek, w którym zidentyfikowano mutację V617 genu JAK2. Praca ta jest częścią wielodyscyplinarnego projektu realizowanego przez Katedrę i Klinikę Hematologii Hospital de Clínicas, mającego na celu określenie obecności tej mutacji u 100 pacjentów z BCR-ABL-ujemnym PMS.
Ostatnia identyfikacja aktywującej mutacji V617F w genie JAK2 stanowi istotny postęp w wiedzy na temat patogenezy BCR-ABL-ujemnych zespołów mieloproliferacyjnych. Ze względu na dużą częstość występowania tej mutacji w PV oraz w mniejszym stopniu w ET i IM postuluje się, że jej wykrycie miałoby duży wpływ na diagnostykę, klasyfikację i leczenie tych jednostek chorobowych. Różne grupy proponowały włączenie wykrywania tej mutacji jako testu diagnostycznego pierwszego rzutu, gdy hematokryt jest większy niż 51% lub gdy podejrzewa się PV, biorąc pod uwagę, że wykrycie JAK2 V617F ma 100% pozytywną wartość predykcyjną dla rozpoznania PV(8,9). Z drugiej strony, kilku autorów postuluje, że obecność tej mutacji pozwoliłaby na nową klasyfikację PMS, ponieważ ET, PV i MI będące nosicielami mutacji JAK2 mogłyby reprezentować tę samą chorobę na różnych etapach ewolucji, a nie różne jednostki(10-12). Jednak dokładna wartość kliniczna i korzyść z wykrycia tej mutacji nie została jeszcze ustalona i wymaga specjalnie zaprojektowanych prospektywnych badań klinicznych, aby odpowiedzieć na te pytania.
Dodatkowo, odkrycie tej mutacji otwiera nowe perspektywy dla terapii ukierunkowanych na zmutowaną kinazę białkową, podobnie jak w przypadku inhibitorów BCR-ABL w przewlekłych białaczkach szpikowych.
Podsumowanie
Polycythemia vera (PV), trombocytemia zasadnicza (ET-TE) i idiopatyczna mielofibroza (IM-MI) są klonalnymi chorobami mieloproliferacyjnymi charakteryzującymi się nadmierną proliferacją jednej lub więcej linii mieloidalnych, takich jak erytrocyty, płytki krwi i fibroblasty szpiku kostnego.
Precyzyjna kategoryzacja zespołów mieloproliferacyjnych nadal wymaga dyskusji, nawet jeśli kryteria diagnostyczne są ścisłe; dodatkowo zaburzenia te są trudne do odróżnienia od procesów reaktywnych.
Ostatnio, w 2005 roku, w wielu z tych jednostek zidentyfikowano mutację JAK2. Sekwencjonowanie regionu kodującego JAK2 ujawniło transwersję G do T w pozycji 1849, która zmieniła walinę na fenyloalaninę (JAK2 V617F).
Częstość występowania mutacji V617F-JAK2 wynosiła prawie 90% u pacjentów z PV i 50% u pacjentów z IM i ET.
W niniejszej pracy opisujemy wykrycie mutacji V617F-JAK2 u pacjenta podejrzanego o PV przy użyciu analizy specyficznej dla alleli reakcji łańcuchowej polimerazy (PCR) oraz omawiamy znaczenie tej mutacji dla diagnostyki i leczenia ujemnych zespołów mieloproliferacyjnych BCR-ABL.
Résumé
La polycytémie vera (PV), Trombocytemia esencjonalna (ET) i idiopatyczna mielofibroza (IM) są blisko spokrewnionymi klonalnymi chorobami mieloproliferacyjnymi charakteryzującymi się nadmierną proliferacją jednej lub więcej linii mieloidalnych, takich jak erytrocyty, płytki krwi i fibroblasty szpiku kostnego.
Chociaż istnieją ścisłe kryteria rozpoznania tych zespołów mieloproliferacyjnych, dokładna kategoryzacja pozostaje kwestią dyskusyjną, co więcej, zaburzenia te są trudne do odróżnienia od procesów reaktywnych w wielu przypadkach.
Ostatnio, w 2005 roku, zidentyfikowano mutację w genie kinazy tyrozynowej Janus kinase 2 (JAK2) w kilku z tych jednostek. Mutacja ta polega na zamianie G na T w pozycji 1849, co skutkuje zamianą w białku fenyloalaniny na walinę (JAK2 V617F).
Częstość występowania tej mutacji stwierdzono w prawie 90% przypadków z PV i w około 50% przypadków z MI i TE.
W niniejszej pracy opiszemy wykrycie tej mutacji u pacjenta z rozpoznaniem prawdopodobnej PV za pomocą specyficznego dla danego allelu testu łańcuchowej reakcji polimerazy (PCR) o wysokiej czułości w wykrywaniu tej mutacji oraz omówimy znaczenie tej nowo odkrytej mutacji w diagnostyce i leczeniu BCR-Zespoły mieloproliferacyjne ABL ujemne.
Resumo
Policitemia Vera (PV), trombocitemia hemorrágica (TE) i mielofibroza idiopática (MI) to klonalne choroby mieloproliferacyjne o silnym związku, charakteryzujące się nadmierną proliferacją jednej lub większej liczby komórek mieloproliferacyjnych, takich jak erytrocyty, płytki i fibroblasty błony śluzowej.
Mimo istnienia rygorystycznych kryteriów rozpoznawania zespołów mieloproliferacyjnych, ich precyzyjna klasyfikacja jest ciągle dyskutowana, a ponadto często bardzo trudno jest odróżnić te zespoły od procesów reaktywnych.
W 2005 roku w kilku z tych jednostek zidentyfikowano mutację w genie kinazy tyrozynowej Janus kinase 2 (JAK2). W mutacji tej obserwuje się substytucję G do T w pozycji 1849, co prowadzi do zamiany fenyloalaniny na walinę (JAK2 V617F) w białku.
Mutację tę obserwowano w około 90% przypadków z PV i w około 50% przypadków z MI i TE.
W niniejszej pracy opisujemy wykrycie tej mutacji u pacjenta z prawdopodobnym rozpoznaniem PV przy użyciu wysoce czułego, specyficznego dla alleli testu łańcuchowej reakcji polimerazy (PCR) do wykrywania tej mutacji i omawiamy znaczenie tej nowo odkrytej mutacji w diagnostyce i leczeniu mieloproliferacyjnych zespołów BCR-ABL-ujemnych.
Bibliografia
1. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054-61.
2. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Layout C, et al. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythaemia vera. Nature 2005; 434: 1144-8.
3. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain of function mutation in JAK2 is frequently found in patients with myeloproliferative disorders. N Engl J Med 2005; 352: 1779-90.
4. Levine RL, Waldleigh M, Cools J, Ebert BL, Werning G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythaemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387-97.
6. Vainshenker W, Constantinescu SN. A unique activation mutation in JAK2 (V617F) is at the origin of Polycythemia Vera and allows a new classification of myeloproliferative diseases. Hematology Am Soc Hematol Educ Program 2005; 195-200.
7. Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3 ed. New York: Cold Spring Harbor Laboratory, 2001.
8. Tefferi A, Pardanani A. Mutation screening for JAK2V617F: when to order the test and how to interpret the results. Leuk Res 2006; 30(6): 739-44.
9. James C, Delhommeau F, Marzac C, Teyssandier I, Couedic JP, Giraudier S, et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia 2006; 20: 350-3.
10. Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005; 366: 1945-53.
11. Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, Ponziani V, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005; 19: 1847-9.