Neutropenia
A lista de todas as causas potenciais de neutropenia não é curta. A etiologia da neutropenia pode ser conceitualmente vista de duas formas amplas, por mecanismo ou categoria etiológica.
Os mecanismos que causam a neutropenia são variados e não completamente compreendidos. Em muitos casos, a neutropenia ocorre após exposição prolongada a uma droga ou outra substância, resultando na diminuição da produção de neutrófilos pela medula óssea hipoplásica. Isto sugere um efeito tóxico directo das células estaminais. Em outros casos, é necessária a exposição repetida mas intermitente a uma droga ou outra exposição. Isto sugere um mecanismo imunológico, embora esta ideia não tenha sido provada. Em muitas situações clínicas, a exposição exata e sua duração em relação ao início da neutropenia não são conhecidas.
Em vista desta compreensão incompleta dos mecanismos de neutropenia, a classificação por ampla categoria etiológica é mais simples de se reter. Neste esquema, a etiologia da neutropenia pode ser classificada como congênita (hereditária) ou adquirida. Embora esta categorização possa ter utilidade diagnóstica clínica limitada, pode ser útil para separar claramente as causas hereditárias da neutropenia da panóplia de causas adquiridas. No cenário das neutropenias hereditárias, estas desordens podem ser descritas como associadas a neutropenias isoladas ou a outros defeitos, sejam imunes ou fenotípicos.
Muitas desordens hereditárias são devidas a mutações na elastase neutrofílica codificadora do gene, ELA2. Vários alelos estão envolvidos. As mutações mais comuns são as substituições intrônicas que inativam um local de emenda no intron 4. Outros genes além do ELA2 também estão envolvidos. A Tabela abaixo lista algumas das condições genéticas envolvidas; estas são condições incomuns.
Tabela 1. Genetic (Hereditary) Conditions in Agranulocytosis (Open Table in a new window)
|
Syndrome |
Inheritance |
Gene |
Clinical Features |
|
Cyclic neutropenia |
Autosomal dominant |
ELA2 |
Alternate 21-day cycling of neutrophils and monocytes |
|
Kostmann syndrome |
Autosomal recessive |
Unknown |
Stable neutropenia, no MDS or AML |
|
Severe congenital neutropenia |
Autosomal dominant |
ELA2 (35-84%) |
Stable neutropenia, MDS or AML |
|
Autosomal dominant |
GFI1 |
Stable neutropenia, circulating myeloid progenitors, lymphopenia |
|
|
Sex linked |
Wasp |
Neutropenic variant of Wiskott-Aldrich syndrome |
|
|
Autosomal dominant |
G-CSFR |
G-CSF–refractory neutropenia, no AML or MDS |
|
|
Hermansky-Pudlak syndrome type 2 |
Autosomal recessive |
AP3B1 |
Severe congenital neutropenia, platelet dense-body defect, oculocutaneous albinism |
|
Chediak-Higashi syndrome |
Autosomal recessive |
LYST |
Neutropenia, oculocutaneous albinism, giant lysosomes, impaired platelet function |
|
Barth syndrome |
Sex linked |
TAZ |
Neutropenia, often cyclic; cardiomyopathy, methylglutaconic aciduria |
|
Cohen syndrome |
Autosomal recessive |
COH1 |
Neutropenia, mental retardation, dysmorphism |
|
Source: Modified from Berliner et al, 2004. AML = acute myeloid leukemia; G-CSF = granulocyte colony-stimulating factor; MDS = myelodysplastic syndrome. |
|||
As causas de neutropenia adquirida são complexas, mas a maioria está relacionada a três categorias principais: infecção, drogas (tanto tóxicas diretas ou imunes mediadas), e auto-imunidade. A neutropenia benigna crônica, ou neutropenia idiopática crônica, parece ser uma desordem de sobreposição com formas hereditárias e adquiridas, e às vezes é indistinguível. Alguns pacientes neutropenos dão uma história clara e padrão familiar, enquanto outros não têm história familiar, poucas determinações de testes sanguíneos e uma duração desconhecida da neutropenia. Este grupo de pacientes pode ter neutropenia hereditária ou adquirida. Segue-se um breve resumo das desordens neutropenianas congênitas e adquiridas.
Neutropenia congênita com defeitos imunológicos associados
Neutropenia com imunoglobulinas anormais é observada em indivíduos com agamaglobulinemia ligada ao X, deficiência de imunoglobulina A (IgA) isolada, síndrome de hiperimunoglobulina M (XHIGM) ligada ao X e disgamaglobulinemia tipo I. No XHIGM, que se deve a mutações no ligante CD40, os pacientes podem realmente ter níveis normais ou elevados de IgM, mas níveis séricos de IgG acentuadamente diminuídos. Em todos esses distúrbios, o risco de infecção é alto, e o tratamento é a imunoglobulina intravenosa (IVIG).
Patientes com disgênese reticular demonstram neutropenia grave, sem imunidade mediada por células, agamaglobulinemia e linfopenia. Ocorrem infecções com risco de vida que são refratárias ao fator de estimulação da colônia de granulócitos (G-CSF). O transplante de medula óssea é o tratamento de escolha.
Congenital ou neutropenias crônicas
Nutropenia congênita severa (SCN), ou síndrome de Kostmann, é mais freqüentemente causada por uma herança recessiva e encontrada em populações remotas e isoladas com alto grau de consanguinidade. Casos autossômicos dominantes e esporádicos também têm sido relatados, na maioria das vezes devido a mutações no receptor do G-CSF. Não existe um defeito genético uniforme nesta síndrome. Mutações no ELA2, que são causadoras de neutropenia cíclica (ver abaixo) não são suficientes para explicar o fenótipo do SCN tipo Kostmann.
Patientes presentes por volta dos 3 meses de idade com infecções bacterianas recorrentes. A boca e o perirectum são os locais mais comuns de infecção. Este tipo de neutropenia é grave, e o tratamento é o G-CSF. O risco de conversão para a síndrome mielodisplásica (MDS)/leucemia mielóide aguda (LMA) com monossomia 7 após o tratamento com G-CSF está associado a mutações adicionais adquiridas. A maioria destes casos é causada por uma mutação no receptor do G-CSF. Pacientes cuja condição responde clinicamente ao G-CSF são tratados para toda a vida.
Alguns pacientes com outras formas de SCN parecem ter mutações no GFI1, um gene repressor transcripcional do dedo de zinco envolvido na função das células-tronco hematopoéticas e decisões de comprometimento da linhagem.
Nutropenia cíclica (CN) é caracterizada por episódios periódicos de neutropenia associada à infecção, seguidos pela recuperação da contagem periférica de neutrófilos. A sua periodicidade é de cerca de 21 dias (intervalo, 12-35 d). Os precursores de granulócitos desaparecem da medula antes de cada nadir de neutrófilos no ciclo devido à apoptose acelerada das células progenitoras mielóides. Alguns casos podem ser determinados geneticamente com uma herança autossômica recessiva. Outros casos podem ser devido a uma herança autossômica dominante. Em alguns casos esporádicos de CN, os pacientes têm mutações no ELA2.
Pessoas com CN tipicamente presentes em bebês ou crianças, mas existem formas adquiridas de CN na vida adulta. O prognóstico é bom, com um curso benigno; no entanto, 10% dos pacientes experimentarão infecções com risco de vida. O tratamento para neutropenia cíclica é G-CSF.
Nutropenia benigna cronicamente
Nutropenia benigna crônica familiar, ou neutropenia étnica benigna, é uma desordem com um padrão autossômico dominante de herança observada em descendência africana, judaica iemenita, judaica etíope, árabe, caribenha e indiana ocidental. Em populações de ascendência judaica africana e iemenita, os estudos genéticos mostram uma forte associação com um polimorfismo uninucleotídeo no gene DARC. Os pacientes são tipicamente assintomáticos, e as infecções são leves. Os indivíduos afetados com neutropenia benigna crônica têm um risco geral baixo de infecção e nenhuma terapia específica é necessária.
Em neutropenias benignas crónicas não-familiares, as infecções leves com um curso benigno tipificam esta desordem. O ANC, entretanto, responde ao estresse, como infecção, corticosteroides e catecolaminas.
Nutropenia crônica grave idiopática
Nutropenia crônica grave idiopática é um diagnóstico de exclusão. Os pacientes afetados apresentam infecções e neutropenia severa.
Neutropenia associada a anormalidades fenotípicas
Shwachman (Shwachman-Diamond) tem um padrão de herança autossômico recessivo. A neutropenia é moderada a grave, com uma taxa de mortalidade de 15-25%, e a síndrome se apresenta na infância, com infecções recorrentes, diarréia e dificuldade de alimentação. Podem ocorrer nanismo, condrodisplasia e insuficiência exócrina pancreática.
Shwachman-Diamond syndrome and X-linked dyskeratosis congenita (DC), cartilage-hair hypoplasia (CHH), e anemia Diamond-Blackfan (DBA), todos parecem compartilhar defeitos genéticos comuns envolvidos na síntese de ribossomos. A maioria dos casos de síndrome de Shwachman-Diamond é causada por mutações no gene da SBDS. A função precisa desse gene ainda está sendo elucidada; no entanto, ele está envolvido na síntese de ribossomos e reações de processamento de RNA. O tratamento é G-CSF.
No CHH, o padrão de herança é autossômico recessivo no cromossomo 9, e é observado nas famílias Amish e Finlandesa. O CHH é causado por mutações no gene RMRP, que codifica o componente RNA do complexo de processamento do RNA mitocondrial ribonuclease (RNase MRP). A neutropenia é moderada a grave. O CHH apresenta defeitos de imunidade mediados por células, anemia macrocítica, doença gastrointestinal e nanismo. Também mostra uma predisposição para o câncer, especialmente linfoma. O tratamento é o transplante de medula óssea.
Disqueratose congênita (síndrome de Zinsser-Cole-Engman) apresenta-se com retardo mental, pancitopenia e imunidade celular mediada por células defeituosas. A disqueratose congênita é mais comum em homens do que em mulheres e é hematologicamente semelhante à anemia de Fanconi. A disqueratose congênita é geralmente recessiva ligada ao X, embora existam formas autossômicas dominantes e autossômicas recessivas desta desordem.
A forma recessiva ligada ao X da desordem tem sido ligada a mutações na DKC1, que codifica a disqueratina, uma proteína nucleolar associada a partículas de ribonucleoproteínas. A forma autossômica dominante está associada a mutações em outro gene, TERC, que é parte da telomerase. A telomerase tem um componente proteico e RNA, e o TERC codifica o componente RNA. Os pacientes com esta doença têm telómeros mais curtos do que o normal. O tratamento é G-CSF, fator estimulante da colônia de granulócitos-macrófagos (GM-CSF), e transplante de medula óssea.
Síndrome de Barth é um distúrbio recessivo ligado ao X que apresenta cardiomiopatia na infância, miopatia esquelética, infecções recorrentes, nanismo, e neutropenia moderada a grave.
Síndrome de Cediak-Higashi é um distúrbio autossômico recessivo com infecções recorrentes, retardamento mental, fotofobia, nistagmo, albinismo oculocutâneo, neuropatia, distúrbios hemorrágicos, gengivite e grânulos lisossômicos em várias células. A neutropenia é moderada a grave e o tratamento é o transplante de medula óssea.
Mielokathexis
Mielokathexis apresenta-se na infância com neutropenia moderada e está associada a infecções recorrentes. A condição é devida a apoptose acelerada e diminuição da expressão de bcl-x em precursores neutrófilos. Observa-se um aspecto nuclear anormal, com hipersegmentação com fios nucleares, piknosis e vacuolização citoplasmática. O tratamento é G-CSF e GM-CSF.
Síndrome dos leucócitos preguiçosos
Síndrome dos leucócitos preguiçosos é uma neutropenia grave com motilidade neutrofílica anormal associada. A etiologia é desconhecida, e o tratamento é de suporte na natureza.
Desordens metabólicas
Estas são neutropenias crônicas com ANCs variáveis. They include glycogen storage disease type 1b and various acidemias, such as isovaleric, propionic, and methylmalonic. In glycogen storage disease type 1b, the treatment is G-CSF and GM-CSF.
Acquired neutropenia caused by intrinsic bone marrow disease
Intrinsic bone marrow diseases that may cause neutropenia include the following:
-
Aplastic anemia
-
Hematologic malignancy (eg, leukemia, lymphoma, myelodysplasia, myeloma)
-
Ionizing radiation
-
Tumor infiltration
-
Granulomatous infection
-
Myelofibrosis
Immune-mediated neutropenia
A drug may act as a hapten and induce antibody formation. This mechanism operates in cases due to gold, aminopyrine, and antithyroid drugs. The antibodies destroy the granulocytes and may not require the continued presence of the drug for their action. Alternatively, the drug may form immune complexes that attach to the neutrophils. This mechanism operates with quinidine.
Drug immune-mediated neutropenia may be caused by the following:
-
Aminopyrine
-
Quinidine
-
Cephalosporins
-
Penicillins
-
Sulfonamides
-
Phenothiazines
-
Hydralazine
Other medications have been implicated
Autoimmune neutropenia is the neutrophil analogue of autoimmune hemolytic anemia and of idiopathic thrombocytopenic neutropenia. It should be considered in the absence of any of the common causes. Antineutrophil antibodies have been demonstrated in these patients. Autoimmune neutropenia may be associated with the following:
-
Rheumatoid arthritis (with or without Felty syndrome)
-
Sjögren syndrome
-
Chronic, autoimmune hepatitis
-
Systemic lupus erythematosus
-
Thymoma
-
Goodpasture disease
-
Granulomatosis with polyangiitis (Wegener granulomatosis)
-
Pure red blood cell (RBC) aplasia, in which there is complete disappearance of granulocyte tissue from the bone marrow; pure RBC dysplasia is a rare disorder due to the presence of antibody-mediated, granulocyte-macrophage colony forming unit (GM-CFU) inhibitory activity, and it is often associated with thymoma
-
Transfusion reactions, which can be caused by the surface antigens of neutrophilia; Os receptores de transfusões repetidas de granulócitos podem tornar-se aloimunizados
-
Proliferação de linfócitos granulares grandes ou leucemia
Em neutropenia neonatal isoimune, a mãe produz anticorpos antineutrófilos IgG para antígenos neutrófilos fetais que são reconhecidos como não-próprios. Isto ocorre em 3% dos nascidos vivos. A doença manifesta-se como febre neonatal, infecção do tracto urinário, celulite, pneumonia e septicemia. A duração da neutropenia é tipicamente de 7 semanas.
A neutropenia auto-imune crônica é observada em adultos e não tem predileção pela idade. Até 36% dos doentes exibem anticorpos antineutrófilos séricos, e o curso clínico é normalmente menos severo. Os doentes podem ter esta doença em associação com lúpus eritematoso sistémico, artrite reumatóide, granulomatose de Wegener e hepatite crónica.
Se a neutropenia auto-imune crónica estiver associada a estas doenças, os corticosteróides estão indicados como tratamento. Em neonatos e crianças, esta desordem está associada a um menor risco de infecção e infecções mais brandas envolvendo o ouvido médio, trato gastrointestinal e pele.
Linfocitose T-gama, ou desordem linfoproliferativa, é uma doença clonal de linfócitos CD3+ T ou CD3- células assassinas naturais (NK) que se infiltram na medula óssea e nos tecidos. Também conhecida como leucemia dos linfócitos granulares grandes (LGL-leucemia), a linfocitose T-gama pode estar associada à artrite reumatóide e está associada a anticorpos antineutrófilos de alto título. A neutropenia é persistente e grave. O tratamento é frequentemente de suporte por natureza, mas também é dirigido para eliminar a população clonal.
Nutropenia adquirida causada por infecção
Infecções são a forma mais comum de neutropenia adquirida. As infecções que podem causar neutropenia incluem, mas não se limitam, às seguintes:
-
Bacterial sepsis
-
Viral infections (eg, influenza, measles, Epstein Barr virus , cytomegalovirus , viral hepatitis, human immunodeficiency virus -1) (see first image below)
-
Toxoplasmosis
-
Brucellosis
-
Typhoid
-
Tuberculosis (see second and third images below)
-
Malaria
-
Dengue fever
-
Rickettsial infection
-
Babesiosis
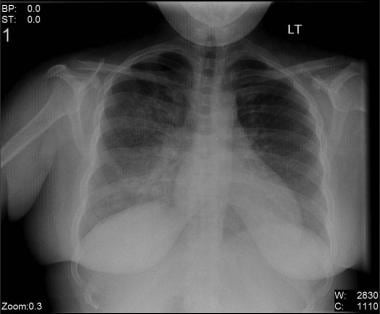 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia. 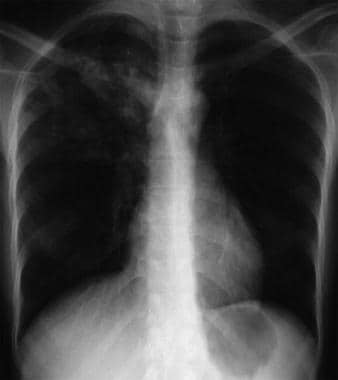 Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mulher foi admitida ao isolamento e iniciou empiricamente um regime de 4 drogas na DE. A tuberculose foi confirmada no exame da expectoração. Imagem cortesia de Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mulher foi admitida ao isolamento e iniciou empiricamente um regime de 4 drogas na DE. A tuberculose foi confirmada no exame da expectoração. Imagem cortesia de Remote Medicine, remotemedicine.org. 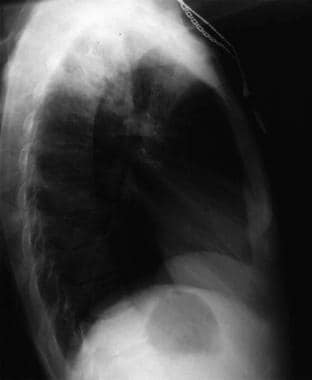 Radiografia de tórax lateral em paciente de 31 anos com pneumonia por influenza. Imagem gentilmente cedida por Remote Medicine, remotemedicine.org.
Radiografia de tórax lateral em paciente de 31 anos com pneumonia por influenza. Imagem gentilmente cedida por Remote Medicine, remotemedicine.org.
 Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.
Bilateral interstitial infiltrates in a 31-year-old patient with influenza pneumonia.  Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mulher foi admitida ao isolamento e iniciou empiricamente um regime de 4 drogas na DE. A tuberculose foi confirmada no exame da expectoração. Imagem cortesia de Remote Medicine, remotemedicine.org.
Anteroposterior chest radiograph in a young ED patient presenting with cough and malaise. The radiograph shows a classic posterior segment right upper lobe density consistent with active tuberculosis. Esta mulher foi admitida ao isolamento e iniciou empiricamente um regime de 4 drogas na DE. A tuberculose foi confirmada no exame da expectoração. Imagem cortesia de Remote Medicine, remotemedicine.org.  Radiografia de tórax lateral em paciente de 31 anos com pneumonia por influenza. Imagem gentilmente cedida por Remote Medicine, remotemedicine.org.
Radiografia de tórax lateral em paciente de 31 anos com pneumonia por influenza. Imagem gentilmente cedida por Remote Medicine, remotemedicine.org. Os organismos mais comumente envolvidos são da flora endógena. Os organismos Staphylococcus aureus são encontrados em casos de infecções cutâneas. Os organismos gram-negativos são observados nas infecções das vias urinárias e gastrointestinais, particularmente Escherichia coli e espécies de Pseudomonas. Também podem ocorrer infecções por Candida albicans. A flora mista pode ser encontrada na cavidade bucal.
Infecções virais levam frequentemente a neutropenia leve ou moderada. A agranulocitose é incomum, mas pode ocorrer. Os organismos mais comuns são o vírus Epstein-Barr, o vírus da hepatite B, o vírus da febre amarela, o citomegalovírus, e a gripe. Muitas infecções, tanto virais como bacterianas, podem causar neutropenia grave.
Nutropenia adquirida causada por deficiência nutricional
Déficiências nutricionais que podem causar neutropenia incluem deficiência de vitamina B-12, folato e cobre.
Nutropenia adquirida causada por drogas e produtos químicos, excluindo quimioterapia citotóxica
Numerosos medicamentos têm sido associados à neutropenia. As categorias de maior risco são medicamentos antitiróides, macrolídeos e procainamidas. Como foi dito acima, muitos medicamentos agem por um mecanismo imuno-mediato. No entanto, alguns medicamentos parecem ter efeitos tóxicos directos nas células estaminais da medula óssea ou nos precursores dos neutrófilos no compartimento mitótico. Por exemplo, drogas como os antipsicóticos e antidepressivos e o cloranfenicol podem actuar como toxinas directas em alguns indivíduos, com base no metabolismo e sensibilidade desta forma. Outros medicamentos podem ter uma combinação de mecanismos imunes e não imunes ou podem ter mecanismos de acção desconhecidos.
Antimicrobianos incluem penicilina, cefalosporinas, vancomicina, cloranfenicol, gentamicina, clindamicina, doxiciclina, flucitosina, nitrofurantoina, novobiocina, minociclina, griseofulvina, lincomicina, metronidazol, rifampicina, isoniazida, estreptomicina, tiacetazona, mebendazol, pirimetamina, levamisole, ristocetina, sulfonamidas, cloroquina, hidroxicloroquina, quinacrina, etambutol, dapsona, ciprofloxacina, trimetoprim, imipenem/cilastatina, zidovudina, fludarabina, aciclovir e terbinafina.
Analgésicos e anti-inflamatórios incluem aminopirina, dipirona, indometacina, ibuprofeno, ácido acetilsalicílico, diflunisal, sulindac, tolmetina, benoxaprofeno, barbitúricos, mesalazina, e quinina.
Antipsicóticos, antidepressivos, e agentes neurofarmacológicos incluem fenotiazinas (clorpromazina, metilpromazina, mepazina, promazina, tioridazina), proclorperazina, trifluoperazina, trimeprazina), clozapina, risperidona, imipramina, desipramina, diazepam, clordiazepoxida, amoxapina, meprobamato, tiotixeno e haloperidol.
Anticonvulsivos incluem ácido valpróico, fenitoína, trimetadiona, mefenitoína (Mesantoína), etosuximida, e carbamazepina.
Antitititireóides incluem tiouracil, propiltiouracil, metimazol, carbimazol, perclorato de potássio, e tiocianato.
As drogas cardiovasculares incluem procainamida, captopril, aprindina, propranolol, hidralazina, metildopa, quinidina, diazoxide, nifedipina, propafenona, ticlopidina, e vesnarinona.
Anti-histaminas incluem cimetidina, ranitidina, tripelennamina (piribenzamina), metafenileno, thenalidina, brompheniramina, e mianserina.
Diuretos incluem acetazolamida, bumetanida, clorotiazida, hidroclorotiazida, clortalidona, methazolamida, e espironolactona.
Os agentes tipoglicêmicos incluem clorpropamida e tolbutamida.
As drogas antimaláricas incluem amodiaquina, dapsona, hidroxicloroquina, pirimetamina e quinina.
Drogas diversas incluem alopurinol, colchicina, aminoglutetimida, famotidina, bezafibrato, flutamida, tamoxifeno, penicilamina, ácido retinóico, metoclopramida, fenindiona, dinitrofenol, ácido etacrílico, diclorodifeniltricloroetano (DDT), cinchophen, antimônio, pirithyldione, rauwolfia, etanol, clorpropamida, tolbutamida, tiazidas, espironolactona, methazolamida, acetazolamida, IVIG e levodopa.
Os metais pesados incluem ouro, arsênico e mercúrio.
Exposição a drogas ou produtos químicos é a causa mais comum de agranulocitose: cerca de metade dos pacientes tem histórico de medicação ou exposição a produtos químicos. Qualquer produto químico ou droga que possa deprimir a medula óssea e causar hipoplasia ou aplasia é capaz de causar agranulocitose. Alguns medicamentos fazem isto a todos se forem administrados em doses suficientemente grandes. Outros agentes parecem causar reações idiossincráticas que afetam apenas certos indivíduos suscetíveis.
Alguns agentes (por exemplo, ácido valpróico, carbamazepina e antibióticos beta-lactam) agem por inibição direta da mielopoiese. Em culturas de medula óssea, esses agentes inibem a formação de colônias de granulócitos de uma forma relacionada à dosagem. O dano direto ao microambiente da medula óssea ou aos precursores mielóides desempenha um papel na maioria dos outros casos.
Muitas drogas associadas à agranulocitose têm sido relatadas à Administração de Alimentos e Medicamentos dos EUA (FDA) sob sua exigência de relato de reações adversas. Muitos agentes também são relatados a um registro mantido pela Associação Médica Americana (AMA). As drogas relatadas foram utilizadas isoladamente, em combinação com outra droga conhecida como potencialmente tóxica, ou com outra droga sem toxicidade conhecida. Vários medicamentos são particularmente salientes devido à sua alta frequência de associação com agranulocitose. Eles incluem o seguinte:
-
Phenothiazine
-
Antithyroid drugs (thiouracil and propylthiouracil)
-
Aminopyrine
-
Chloramphenicol
-
Sulfonamides
Miscellaneous immunologic neutropenias
Immunologic neutropenias may occur after bone marrow transplantation and blood product transfusions.
Felty syndrome is a syndrome of rheumatoid arthritis, splenomegaly, and neutropenia. Splenectomy shows an initial response, but neutropenia may recur in 10-20% of patients. Treatment is directed toward rheumatoid arthritis.
In complement activation–mediated neutropenia, hemodialysis, cardiopulmonary bypass, and extracorporeal membrane oxygenation (ECMO) expose blood to artificial membranes and can cause complement activation with subsequent neutropenia.
In splenic sequestration, the degree of neutropenia resulting from this process is proportional to the severity of the splenomegaly and the bone marrow’s ability to compensate for the reduction in circulating bands and neutrophils.
Eosinopenia and basophilopenia
Eosinopenia may be associated with the following:
-
Acute bacterial infection
-
Glucocorticoid administration
-
Physical stress
-
Thymoma
Decreased circulating basophils may be associated with the following:
-
Anaphylaxis
-
Acute infection
-
Drug-induced hypersensitivity
-
Congenital absence of basophils
-
Hemorrhage
-
Hyperthyroidism
-
Ionizing radiation
-
Neoplasia
-
Ovulation
-
Urticaria
-
Drugs (eg, corticosteroid, adrenocorticotropic hormone therapy, chemotherapeutic agents, thyroid hormones)
Go to Pediatric Autoimmune and Chronic Benign Neutropenia for complete information on this topic.